β- 酮硫解酶缺乏症:病因与流行病学、临床表现、诊断和治疗
2022-09-02 MedSci原创 MedSci原创

β-酮硫解酶缺乏症(beta-ketothiolase deficiency,BKD)又称线粒体乙酰乙酰基辅酶A硫解酶缺乏症,是一种罕见的常染色体隐性遗传代谢病。患者线粒体内异亮氨酸代谢障碍
β-酮硫解酶缺乏症(beta-ketothiolase deficiency,BKD)又称线粒体乙酰乙酰基辅酶A硫解酶缺乏症,是一种罕见的常染色体隐性遗传代谢病。患者线粒体内异亮氨酸代谢障碍,肝外酮体分解障碍,急性期出现酮症低血糖、代谢性酸中毒,严重者猝死或残障,经过饮食干预可以有效控制。
一、病因和流行病学
β- 酮硫解酶在异亮氨酸代谢和酮体分解过程中发挥重要作用,它负责催化异亮氨酸代谢过程的第 6 步由 2- 甲基 - 乙酰乙酰基辅酶 A 分解为乙酰辅酶 A 和丙酰辅酶 A,同时也在酮体分解和脂肪酸氧化过程中催化乙酰乙酰辅酶A 生成乙酰辅酶 A。乙酰辅酶 A 乙酰基转移酶 -1(ACAT1) 基因突变是导致 BKD 的病因。当 ACAT1 基因突变引起酶活性缺陷时,异亮氨酸的正常分解代谢和肝外酮体利用受到阻滞,致使大量酸性中间代谢产物和酮体在血液、组织中积聚,表现为代谢性酸中毒及酮症。
ACAT1 基因定位于 11q22.3,基因全长约 27kb,包含 12 个外显子及 11 个内含子,在其 cDNA 翻译后加工及修饰形成包含 394 个氨基酸的 β- 酮硫解酶。自 1971 年第 1 例 BKD 患者被报道以来,ACAT1 基因已报道 104 种突变,类型包括错义突变、无义突变、剪切突变等;多数是单个核苷酸变异,也有 DNA 片段的缺失及重复报道。国内 6 例 BKD 报道, 发现 4 个已报道变异及 2 个新变异,均属错义突变,目前尚无热点突变的详细报道。
二、流行病学
该病的发病率为 1/333 000~1/111 000,不同国家和地区存在较大差异。中国浙江省在对 1 861 262 名新生儿遗传代谢病串联质谱技术筛查,发现 2 例诊断为β- 酮硫解酶缺乏症, 发病率为 1/960 600。
β-酮硫解酶缺乏症是一种罕见的常染色体隐性遗传代谢疾病,全世界仅报告有50至60例。患者机体无法正确处理异亮氨酸或脂质分解产物,典型发作年龄为6个月至24个月。
三、临床表现
婴儿及儿童期起病。首次发作时年龄在 5 个月至 7 岁。临床表现为反复发作的难治性呕吐及重症酮症酸中毒,伴有萎靡、脱水、呼吸急促、昏迷等临床表现。通常有明确诱因,可在禁食、胃肠道及上呼吸道感染、发热、应激或过量摄入蛋白质后急性起病。常规的实验室检查可见血糖升高或正常,部分出现低血糖,血气显示代谢性酸中毒,血氨可以正常或轻度升高,肝功能、肾功能及正常,尿酮体阳性。酮症及酸中毒可反复出现,发作间期临床及实验室检查可以表现为正常。
约 1/5 的病例出现神经系统的改变,表现为认知障碍、智力发育落后及锥体外系异常及脑卒中的症状,头颅 MRI 可发现包括白质广泛的脱髓鞘,基底节区在 T2 可见对称性高信号改变。
患者早期可无症状,在新生儿期或高危人群通过血、尿代谢筛查时被诊断。起病年龄、发作频率与预后无明确关联。发作随年龄增长有减少或停止趋势,多数病例随访其生长发育未受影响。反复发作及严重者可致多器官功能障碍甚至死亡。
四、诊断
对于婴幼儿期酮症酸中毒的患者,应高度警惕,及早进行尿有机酸分析、血氨基酸及酯酰肉碱谱分析、基因检测,患者特征性表现为尿2-甲基3-羟基丁酸、甲基巴豆酰甘氨酸、乙酰乙酸增高,血液3-羟基异戊酰肉碱、乙酰肉碱增高,游离肉碱降低,ACAT1基因检出纯合或复合杂合突变可获得分析诊断。
1. 临床上有反复发作的难治性呕吐合并酮症酸中毒者要警惕该病。
2. 通常有明确诱因,可在禁食、胃肠道及上呼吸道感染、发热、应激或过量摄入蛋白质后急性起病。
3. 常规检测可见尿酮体阳性,血气分析 pH 降低,部分患者可有血糖明显升高或降低,血氨浓度正常或升高。血常规及肝功能多无明显异常。
4. ①血串联质谱检测酰基肉碱谱可见血 3- 羟基戊酰肉碱(C5OH)、3- 羟基丁酰肉碱(C4OH) 及异戊烯酰肉碱(C5 :1) 浓度升高;②尿气相色谱质谱检测可见尿 2- 甲基 -3- 羟基丁酸(2M3HB)、甲基巴豆酰甘氨酸(TIG) 及 2- 甲基乙酰乙酸(2MAA) 明显升高;③外周血白细胞及成纤维细胞硫解酶活性明显降低;④基因检测明确有 ACAT1 基因突变。
五、鉴别诊断
1. β- 酮硫解酶缺乏症的临床表现及常规生化检测与 C5OH 增高的其他有机酸血症相似,需要与 3- 甲基巴豆酰辅酶A 羧化酶缺乏症、3- 羟基-3- 甲基戊二酰辅酶A 裂解酶缺乏症相鉴别。鉴别要点是要同时进行血酰基肉碱谱和尿有机酸分析,前者除了 C5OH 升高外,同时伴有 C4OH 和 C5 :1 升高,尿中可检出 2- 甲基 -3- 羟基丁酸、甲基巴豆酰甘氨酸及 3- 羟基丁酸浓度明显升高。
2. 伴有血糖升高时,需与糖尿病酮症酸中毒鉴别。
六、治疗
治疗原则为及时纠正酸中毒,高热量饮食,尽可能减少酮症酸中毒和低血糖发作。
1.急性发作期
静脉滴注葡葡糖、电解质溶液及左卡尼汀等,保证充足的液体入量,促进有机酸类代谢毒物的排泄,给予充足的热量供给,减少蛋白质分解,减少酸性物质的产生。同时,根据个体情况给予碳酸氢盐,严重患者需要血液透析或血浆置换。左卡尼汀可与有机酸代谢产物形成酰基肉碱,有利于有机酸的排泄。解除发热、感染等诱发因素,静脉输入葡萄糖以减少蛋白质持续分解、保证热量供应2.稳定期治疗。
正常进食,少食多餐,避免长时间空腹,避免疲劳,轻微限制蛋白质,以避免摄取过多的异亮氨酸,并需注意避免高脂肪饮食。口服左卡尼汀有助于稳定代谢状况。
另有研究报道胰岛素对 BKD 患者非糖尿病性酮症酸中毒的治疗确切有效。
七、诊疗流程
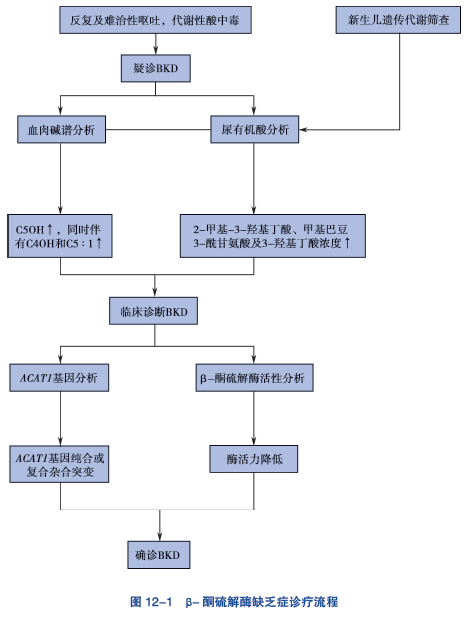
图 12-1 β-酮硫解酶缺乏症诊疗流程
八、预后
首次发作时如能得到及时诊断和合理治疗,多数β-酮硫解酶缺乏症患儿可完全康复,发育正常。如未能及时诊断和治疗,患儿反复发生酸中毒,严重者死亡,幸存者可能遗留严重的神经系统后遗症。因此,及时合理的治疗和长期管理对改善预后十分重要。
九、罕见病信息登记
如果您愿意寻求不断更新的信息,建议您在此登记患者的信息,即使没有完全确诊,也可以登记,点击进入:
参考资料:
Nguyen KN,Abdelkreem E,Colombo R,et al.Characterization and outcome of 41 patients with beta- ketothiolase deficiency:10 years’experience of a medical center in northern Vietnam.J Inherit Metab Dis, 2017,40(3):395-401.
洪芳,黄新文,张玉,等 . 浙江省新生儿有机酸尿症筛查及随访分析 . 浙江大学学报(医学版),2017,46(3):240-247.
https://www.chard.org.cn/#/knowledge/jbzsk/detail/31
https://baike.baidu.com/item/%CE%B2-%E9%85%AE%E7%A1%AB%E8%A7%A3%E9%85%B6%E7%BC%BA%E4%B9%8F%E7%97%87
https://rarediseases.org/gard-rare-disease/beta-ketothiolase-deficiency/
本网站所有内容来源注明为“williamhill asia 医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于williamhill asia 医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“williamhill asia 医学”。其它来源的文章系转载文章,或“williamhill asia 号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与williamhill asia 联系,williamhill asia 将立即进行删除处理。
在此留言




#流行病#
63