
Circ Res 天津医科大学陈雄文教授团队揭示PKA是生理性及病理性心肌肥厚的关键调控因子
2024-01-30 论道心血管 论道心血管 发表于上海

该研究揭示PKA是生理性及病理性心肌肥厚的关键调控因子,为病理性心肌肥厚的防治提供了新策略。
心肌肥厚(cardiac hypertrophy,CH)是在生理性或病理性应激条件下,心脏为满足心输出量需求增加导致的心脏负荷增加而产生的一种适应。生理性CH (physiological CH,PhCH)是在出生后生长期、妊娠期和运动训练期间,在生理性应激条件下,交感-肾上腺素能系统(sympathetic-adrenergic system,SAS)激活导致的短期内心脏大小的适应性增加,它对心脏收缩功能没有负面影响。然而,高血压、心肌梗死或瓣膜功能障碍等病理性应激导致的SAS持续激活则会对心脏产生不良影响。最初的代偿性肥厚过程可演变为病理性CH (pathological CH,PaCH),并导致心脏收缩功能障碍、心肌硬化、间质纤维化,最终导致心力衰竭。预防和治疗PaCH被认为是减少心脏处于病理性应激下的患者心力衰竭和心律失常发病率的重要措施。
蛋白激酶A (protein kinase A,PKA)是心脏中SAS的主要效应分子,通过直接磷酸化下游蛋白效应物或调节特定基因的转录,负责调节心血管稳态并参与心脏疾病的发病过程。持续性β1-肾上腺素能信号刺激导致PKA的过度激活和PaCH的发生。除cAMP外,PKA还可被活性氧、血管紧张素II和内皮素I、脂多糖和炎症因子等非经典激活因子激活,这些因子会在心脏受应激刺激条件下上调。目前研究表明,不同的信号传导通路分别负责调控PhCH和PaCH。介导PhCH的最具特征的信号传导通路是IGF1-PI3K-Akt通路,调控PaCH发生的经典信号传导通路则是GPCR的过度激活,而两种形式的CH都涉及mTORC信号通路的激活。由于SAS的激活参与了PhCH和PaCH, SAS和其主要效应分子PKA的激活可能是这两种形式CH的共同调节信号。由于缺乏PKA敲除动物模型和心脏特异性PKA抑制剂,PKA在CH中的作用仍有争议,其具体分子调控机制仍未阐明。
2024年1月26日,天津医科大学陈雄文教授团队在Circulation Research发表题为“Protein Kinase A Is a Master Regulator of Physiological and Pathological Cardiac Hypertrophy”的研究论文,揭示PKA是生理性及病理性心肌肥厚的关键调控因子,为病理性心肌肥厚的防治提供了新策略。

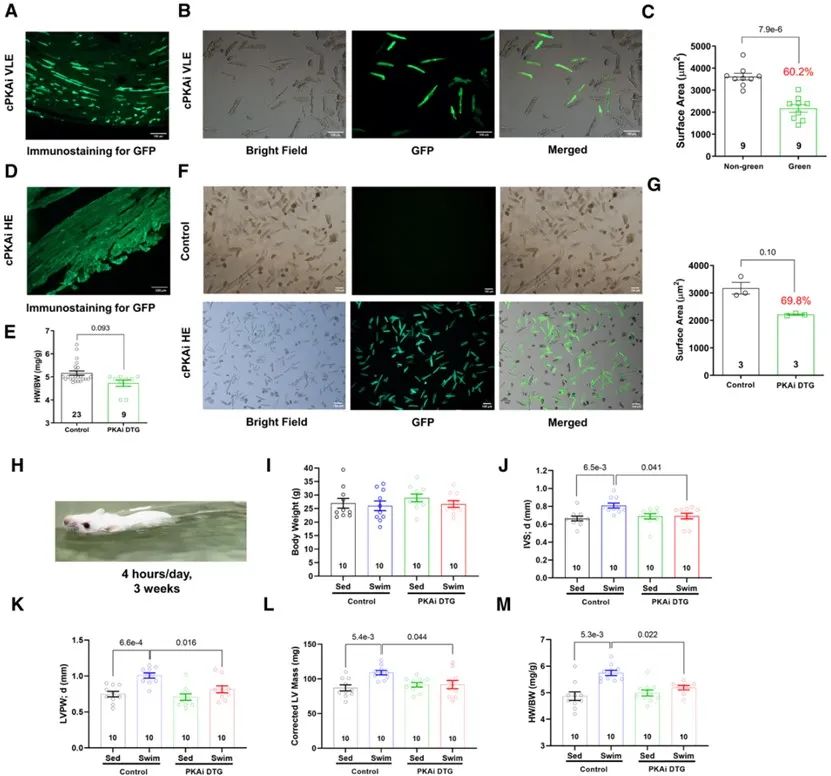
先前的研究表明,PKA抑制肽(PKA inhibition peptide,PKAi)的PKA抑制结构域α (PKIα,PKAi的氨基端第1-25位氨基酸片段)可以高度特异并有效地抑制PKA的活性,其与绿色荧光蛋白(GFP)的融合蛋白能在细胞内稳定存在。为探究PKA在PhCH和PaCH中的作用,研究人员构建了心肌细胞特异性(由αMHC启动子控制)和诱导性(由四环素诱导的反式激活因子控制)的PKAi-GFP过表达的双转基因(DTG)小鼠。PKAi-GFP DTG小鼠在3周龄断奶后停用含多西环素饲料,因插入的拷贝数和插入位点不同导致转录水平不同,研究人员获得了PKAi-GFP高、中、低、极低表达(HE/ME/LE/VLE)的四种DTG小鼠品系,其对于1µM cAMP诱导的PKA活性的最大抑制率分别为95%、57%、20%和10%。研究人员分离4月龄PKAi-GFP VLE DTG小鼠的心肌细胞,发现PKAi-GFP+的心室肌细胞表面积显著小于PKAi-GFP-的心室肌细胞。而与对照小鼠相比,PKAi-GFP HE DTG小鼠的心脏重量/体重比值(HW/BW)和心肌细胞表面积均减小。为研究心肌细胞PKA是否参与运动诱导的PhCH,研究人员给予对照小鼠和PKAi-GFP ME DTG小鼠游泳锻炼3周构建PhCH模型。造模结束时,超声心动图分析显示,运动增加对照小鼠的舒张期室间隔及左室后壁的厚度,并增加了HW/BW,而PKAi-GFP DTG小鼠则没有上述改变。因此,心肌PKAi (cPKAi)延缓了出生后心脏生长,抑制了运动诱导的PhCH。
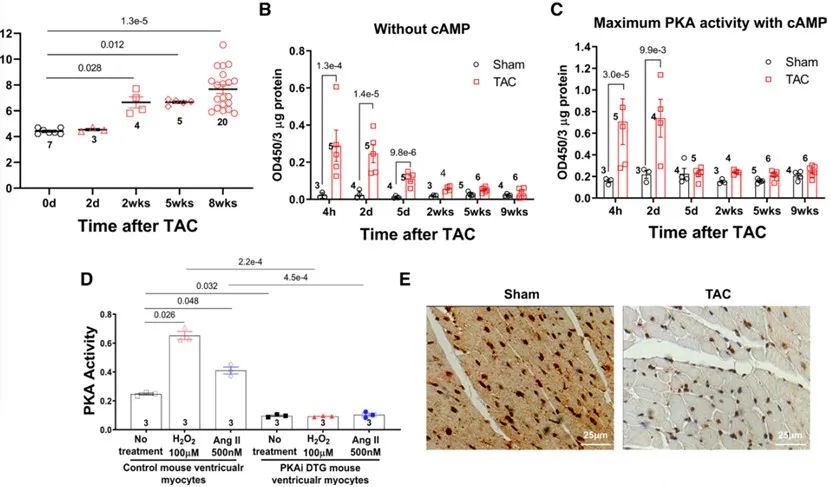
随后,研究人员对野生型小鼠进行胸主动脉缩窄术(TAC)以构建PaCH动物模型。经ELISA测定,TAC组小鼠心脏匀浆中PKA活性在TAC术后2天显著升高,在术后5天回落,在此后,TAC组小鼠心脏PKA活性仍有高于假手术组小鼠的趋势。研究人员以1 μmol/L cAMP诱导TAC小鼠心脏匀浆中PKA的最大活性,结果显示,在术后2天内的TAC组心脏组织PKA活性与假手术组相比显著升高。免疫组织化学结果显示,TAC术后2周小鼠心脏中PKIα的表达显著低于对照组小鼠。此外,非经典的PKA激活因子H2O2和血管紧张素II可以提高对照小鼠心肌细胞PKA活性,但不能激活PKAi-GFP DTG小鼠心肌细胞PKA。以上结果说明,SAS激活、内源性PKIα的缺失以及非经典的PKA激活因子的刺激导致TAC后心脏PKA被激活。
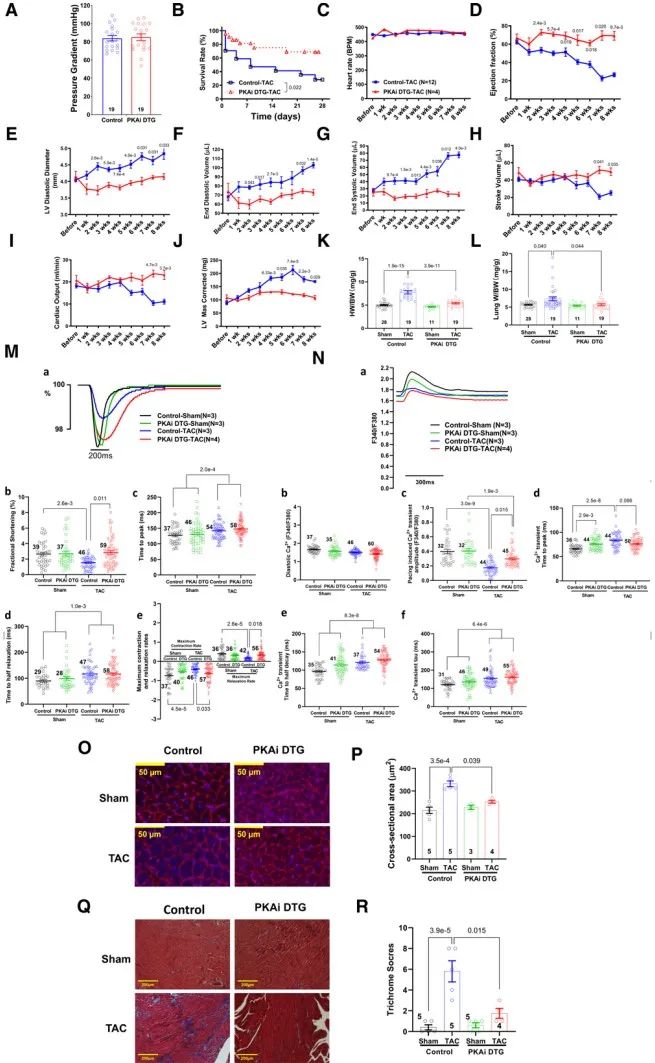
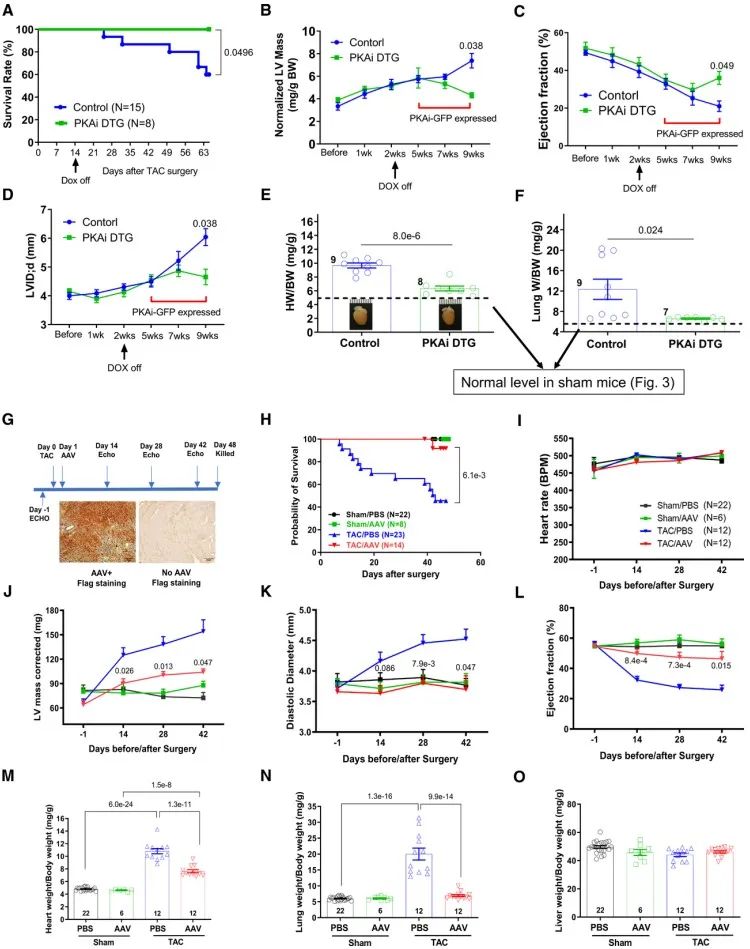
为探究PKA在PaCH中的作用,研究人员对TAC术后8周内的对照小鼠和PKAi-GFP ME DTG小鼠每周进行超声心动图检查,结果显示,TAC可显著降低对照小鼠的每搏输出量、射血分数和心输出量,显著升高左室舒张直径、舒张末期容积、收缩末期容积和左室质量,而在PKAi-GFP DTG小鼠中则无明显改变。血流动力学方面,cPKAi改善了TAC后心脏舒张功能(舒张末压降低,-dp/dt增加)和收缩功能(最大dp/dt增加,收缩指数增加)。TAC术后8周,cPKAi显著降低HW/BW和肺重量/体重比,并显著减少了TAC引起的心肌细胞肥大和间质纤维化。并且,PKAi-GFP DTG小鼠在TAC术后4周内的存活率显著高于对照小鼠。对TAC术后8周的对照小鼠和PKAi-GFP ME DTG小鼠进行收缩功能及钙瞬变检测,结果显示,cPKAi在TAC术后的保护作用可能是通过缓解心肌细胞钙瞬态抑制和心肌收缩力下降来实现的。β-肾上腺素能过度刺激可以通过增强L型Ca2+通道介导的Ca2+内流引起PaCH,而Ca2+内流不受PKA激活调控。为探究cPKAi是否可以缓解Ca2+内流增加所诱导的CH,研究人员将PKAi-GFP TG小鼠与心肌细胞特异性Cavβ2a/ttA DTG小鼠杂交,获得了Cavβ2a/ttA/PKAi TTG小鼠。研究人员发现,Cavβ2a/ttA/PKAi TTG小鼠的生存率显著高于Cavβ2a DTG小鼠。超声心动图结果显示,cPKAi可显著减小舒张末期左室内径,从而减少每搏输出量和心输出量;而cPKAi减小了舒张期左室后壁厚度,导致校正后的左室质量及HW/BW减小。
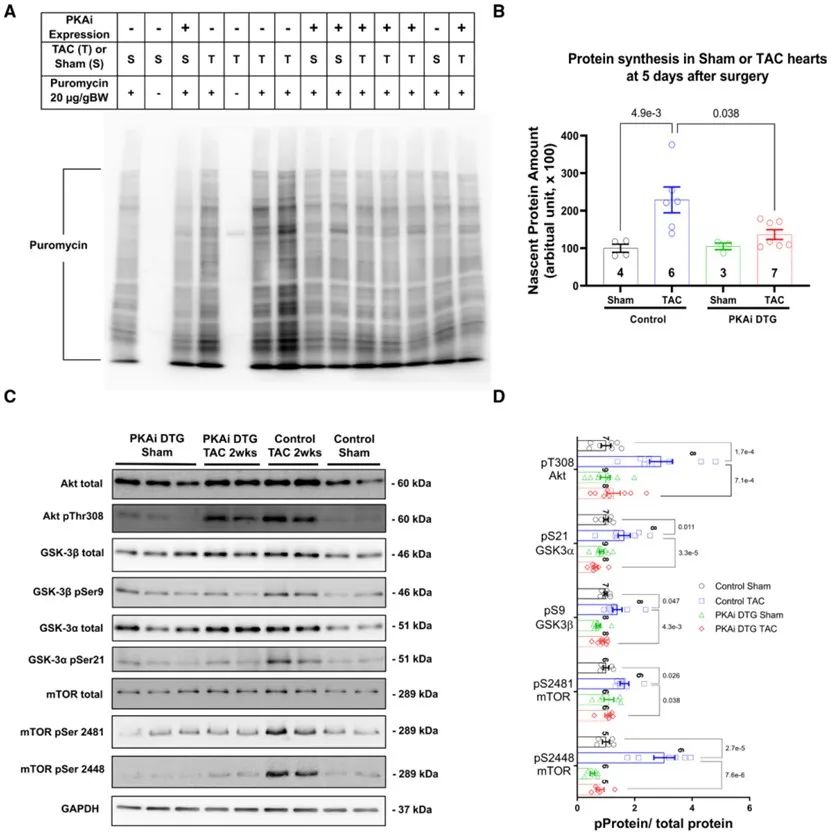
由于蛋白质合成在PaCH的发生发展中至关重要,接下来,研究人员通过检测新生蛋白的合成率来探究cPKAi是否通过抑制蛋白合成来改善PaCH,发现TAC增加了对照组小鼠心脏的蛋白质合成,但在cPKAi DTG小鼠心脏中没有此现象。大量研究表明,mTOR信号通路在蛋白质合成中发挥重要作用,受Akt、GSK-3α、GSK-3β和PKA调控。GSK-3可抑制mTOR活性,而PKA使其磷酸化并失活,从而激活mTOR。Western blot结果表明,TAC诱导mTOR Ser2448和Ser2481位点磷酸化增加,从而使其活化,可能是通过磷酸化并激活Akt和磷酸化GSK-3α、GSK-3β并使其失活导致的。cPKAi逆转了TAC对蛋白质合成信号传导的增强作用。这些发现证明PKA通过增加蛋白合成介导了TAC诱导的PaCH。
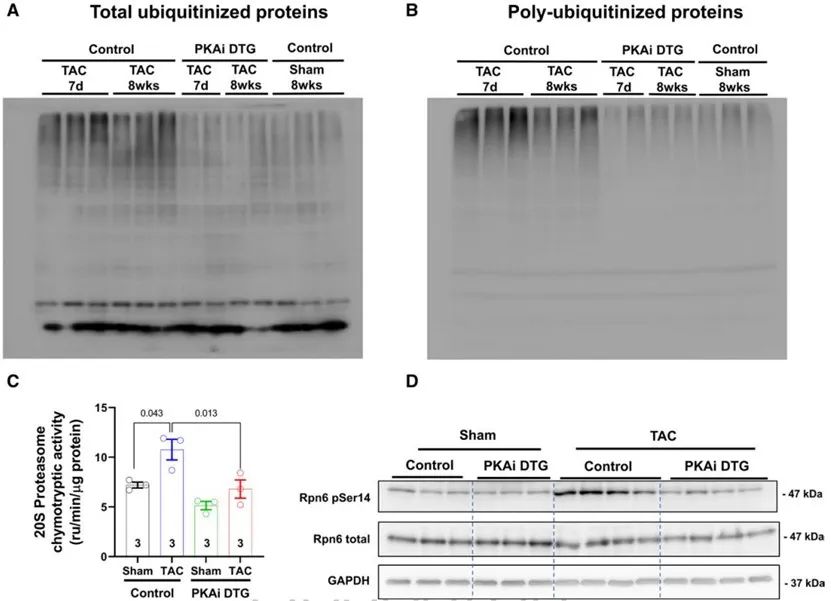
正常情况下,当蛋白质合成增加时,蛋白质降解也会增加,以达到蛋白质稳态。然而,在PaCH中,存在错误折叠蛋白的蓄积。研究人员发现,在TAC术后7天和8周,与假手术组小鼠心脏相比,TAC组小鼠心脏中蛋白质的泛素化和多聚泛素化增多。对照小鼠中,TAC增加了心脏中20S蛋白酶体的活性,而没有增加PKAi-GFP DTG小鼠心脏的20S蛋白酶体活性。PKA介导的RPN6磷酸化对于蛋白酶体活性至关重要, cPKAi可显著抑制TAC引起的小鼠心脏RPN6的磷酸化。以上结果说明,cPKAi通过降低RPN6磷酸化从而降低泛素-蛋白酶体系统对蛋白质的降解。
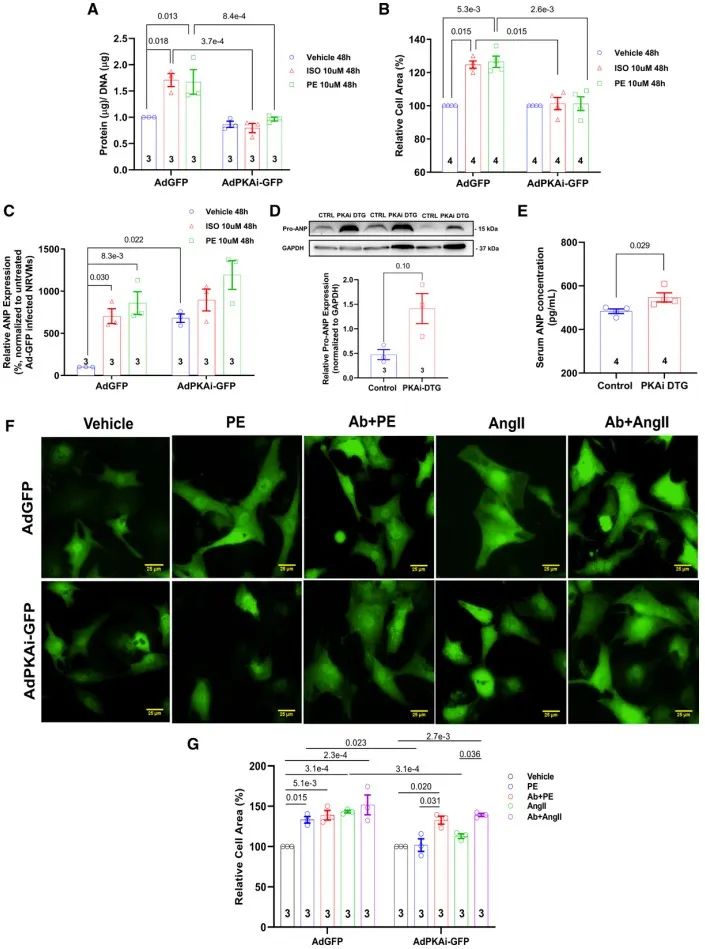
为了在体外水平验证PKA在CH中的作用,研究人员对转染过表达PKAi-GFP腺病毒(AdPKAi-GFP)或对照腺病毒(AdGFP)的新生大鼠心肌细胞(NVRMs)给予异丙肾上腺素(ISO)和苯肾上腺素(PE)刺激。经ISO或PE刺激后,转染AdGFP的细胞表面积和蛋白/DNA比值显著增加,而转染AdPKAi-GFP的NRVMs的表面积和蛋白/DNA比值几乎没有增加。在过表达PKAi-GFP的NRVMs中ANP mRNA表达与对照组相比显著升高,PKAi-GFP DTG小鼠心脏pro-ANP表达及血清中ANP水平在没有病理性刺激的情况下与对照组相比亦显著升高。当用ANP中和抗体中和培养基中的ANP时,PKAi-GFP对PE和血管紧张素II诱导的心肌肥大的抑制作用被消减。这些结果表明,cPKAi抑制NRVMs肥大的作用可能是部分通过上调具有抗肥大作用的ANP的表达来实现的。
以上结果已表明,压力负荷增加前,cPKAi可以预防PaCH。研究人员接下来又进一步明确cPKAi是否在PaCH模型建立后仍具有治疗作用。对TAC术后2周的cPKAi HE DTG小鼠中,去除多西环素喂养,以诱导在PKAi-GFP在TAC术后5周表达。当PKAi-GFP不表达时,TAC术对于对照组小鼠和PKAi-GFP DTG小鼠心脏有相同程度的损害;而诱导PKAi-GFP表达后,PKAi-GFP DTG小鼠较对照组左室质量、舒张末期左室内径显著降低,射血分数显著增高,存活率亦显著升高。研究人员还使用rAAV9介导的cPKAi过表达来验证了上述结果。这些结果均说明,cPKAi可以改善已经发生的PaCH。
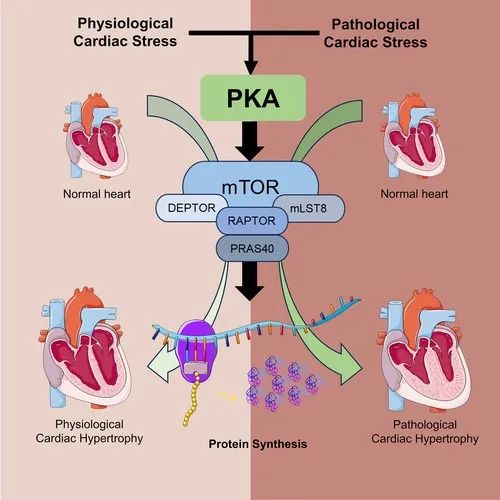
综上所述,PKA通过调节蛋白质合成和降解机制调节出生后生理性心肌细胞生长、运动诱导的PhCH以及压力过载诱导或Ca2+流入增加诱导的PaCH。在心脏应激时,PKA活性可通过SAS激活和非经典的PKA激活因子增强,并伴随心脏内源性PKIα的减少。cPKAi可以阻止PKA过度激活,可以预防并治疗PaCH。选择性抑制PKA可能是改善PaCH和预防继发性心力衰竭及心律失常的新策略。
天津医科大学陈雄文教授及美国天普大学Xiaoying Zhang是该论文的共同通讯作者。天津医科大学白英玉、美国天普大学 Xiaoying Zhang和第二炮兵总医院Ying Li为该论文的共同第一作者。
原文链接:
https://www.ahajournals.org/doi/abs/10.1161/CIRCRESAHA.123.322729
本网站所有内容来源注明为“williamhill asia 医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于williamhill asia 医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“williamhill asia 医学”。其它来源的文章系转载文章,或“williamhill asia 号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与williamhill asia 联系,williamhill asia 将立即进行删除处理。
在此留言










不错,学习了。
43
#心肌肥厚# #PKA#
51