
【佳作推荐】 百度科研团队NMI期刊论文:一种使用蛋白质语言模型进行结构预测的新方法
2023-11-24 ComputArt计算有乐趣 ComputArt计算有乐趣 发表于上海

作者提出了一种无需MSA搜索的蛋白质结构预测方法——HelixFold-Single。
基于人工智能的蛋白质结构预测方法,例如Alphafold2,已经达到了接近实验的准确性。这些方法主要依靠多序列比对(multiple sequence alignments, MSA)作为输入来从若干同源序列中学习共同进化信息。然而,过度依赖MSA也成为各种蛋白质相关任务的瓶颈,耗时的MSA搜索给需要高通量的任务(例如蛋白质设计)带来了巨大的阻碍。为了解决这一问题,作者提出了一种无需MSA搜索的蛋白质结构预测方法——HelixFold-Single,该方法采用大规模蛋白质语言模型(large scale protein language model, PLM)作为 MSA 的替代,以学习无MSA搜索的共同进化知识,而后通过结合预先训练的蛋白质语言模型和AlphaFold2的基本模块得到最终端到端的可微模型,该模型可以仅从一级序列预测蛋白质的三维坐标。近日,该项研究工作发表在Nature Machine Intelligence期刊上。(Nat Mach Intell 2023, 5 (10), 1087–1096)
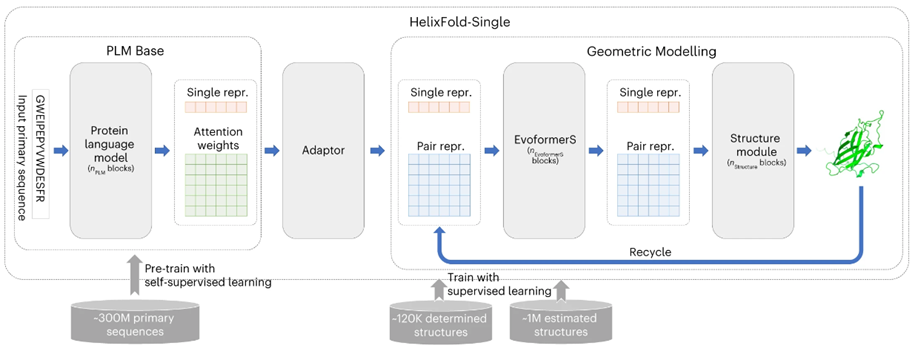
HelixFold-Single的基本架构如图1所示。模型由三个部分组成:PLM Base、Adapter 和 Geometric Modelling。作者首先采用大规模PLM Base对蛋白质序列中的协同进化信息进行编码,作为MSA的替代方案。然后,作者采用适配层从 PLM中提取协同进化信息,以有效生成几何建模输入所需的单序列特征和残基对特征。在几何建模中,作者参考Alphafold2的基本架构,使用修改后的Evoformer(名为EvoformerS)和结构模块来充分交换单个序列特征表示和残基对特征表示之间的信息,以捕获几何信息并恢复原子的3D坐标。
HelixFold-Single的训练由两个阶段组成,在第一阶段,通过掩码语言预测任务,使用数百万个未标记的蛋白质序列训练大规模PLM base。随后在第二阶段,使用带标签的蛋白三维实验结构以及Alphafold2生成结构来训练整个模型。
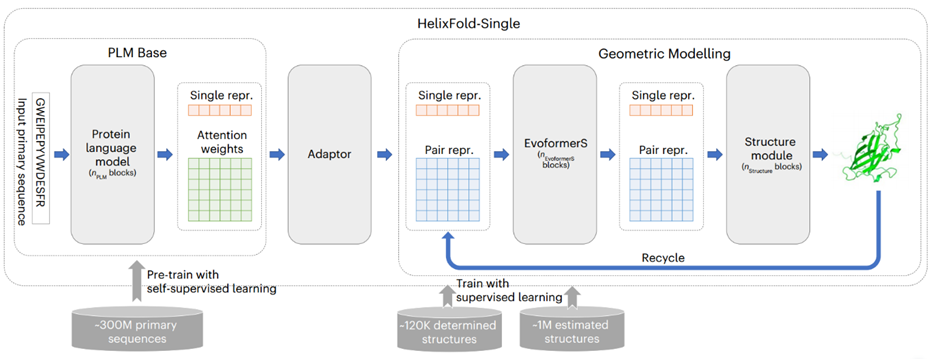
图1、HelixFold-Single模型架构
随后作者将HelixFold-Single与两种主流蛋白质预测模型Alphafold2和RoseTTAFold在CASP14和CAMEO数据集上进行了比较(见图2)。总体而言,HelixFold-Single显著超越了CASP14 和CAMEO 上所有无 MSA 的方法,并且在某些场景下与基于MSA 的方法相当。HelixFold-Single 展示了将 PLM 融入几何建模以进行蛋白质结构预测的潜力。研究人员还分析了HelixFold-Single在具有不同数量同源序列的靶标上的性能,结果表明HelixFold-Single的准确性与同源序列的数量有关,在具有大同源家族的靶点上HelixFold-Single的预测准确性可以与基于 MSA 的方法相媲美。
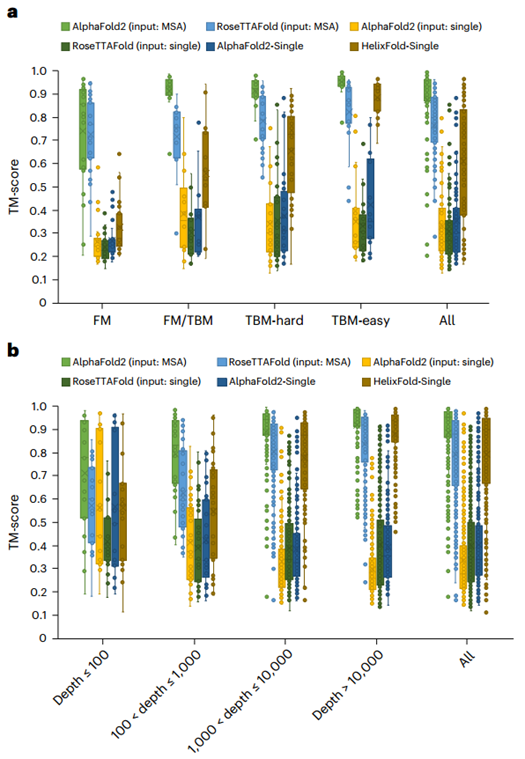
图2、HelixFold-Single与其他方法在CASP14和CAMEO数据集上的总体比较
此外作者还比较了HelixFold-Single与Alphafold2的预测速度(如图3所示)。与基于 MSA 的方法相比,HelixFold-Single的优势在于其效率,因此它非常适合高通量蛋白质结构预测任务,例如蛋白质设计。
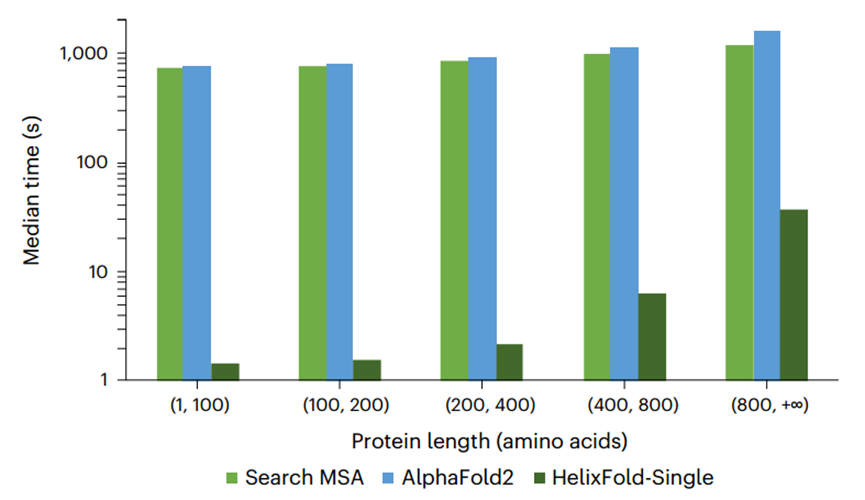
图3、Alphafold2预测、MSA搜索与HelixFold-Single预测的中位数时间比较
在该项研究工作中,为了避免耗时的MSA搜索过程对大规模蛋白质预测的限制,作者提出了一种无需MSA搜索预测蛋白结构的方法HelixFold-Single。HelixFold-Single 尝试利用 PLM 和几何建模的优势,仅通过一维序列来进行端到端的蛋白质结构预测。通过利用蛋白语言模型中的大规模参数嵌入同源信息,作者证明PLM可以作为 MSA 的替代品,以减少蛋白质结构预测所需的时间。HelixFold-Single对于具有大同源家族的靶标可以与基于MSA的方法相媲美,并且比基于MSA的方法高效得多,展示了其在蛋白质研究中的应用前景。有实验结果表明:更大规模的PLM可以实现更优越的性能,后续有望通过使用更大规模的PLM并引入更多样化的数据来进一步提高模型预测蛋白质结构的性能。
参考文献:
(1) Fang, X.; Wang, F.; Liu, L.; He, J.; Lin, D.; Xiang, Y.; Zhu, K.; Zhang, X.; Wu, H.; Li, H.; Song, L. A Method for Multiple-Sequence-Alignment-Free Protein Structure Prediction Using a Protein Language Model. Nat Mach Intell 2023, 5 (10), 1087–1096. https://doi.org/10.1038/s42256-023-00721-6.
本网站所有内容来源注明为“williamhill asia 医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于williamhill asia 医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“williamhill asia 医学”。其它来源的文章系转载文章,或“williamhill asia 号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与williamhill asia 联系,williamhill asia 将立即进行删除处理。
在此留言








#人工智能# #蛋白质结构预测方法# #蛋白质语言模型# #多序列比对#
47