Nat Med:ERK可能是胰腺癌潜在的靶点
2019-04-03 杨冰 iProteome

胰腺癌(PDAC)的肿瘤发生依赖于KRAS和自噬,但是KRAS在自噬方面的作用仍然未知,这篇研究表明KRAS的抑制和ERK MAPK的药理抑制增加了自噬通量,而且削弱了糖酵解和线粒体功能。作者设想抑制ERK可以增强PDAC对自噬的依赖,这可能是通过损害KRAS或ERK驱动的代谢过程。他们发现自噬抑制剂和特异性自噬调节因子的遗传或者药理学抑制,协同增强了ERK抑制剂介导KRAS驱动的PDAC中抗
胰腺癌(PDAC)的肿瘤发生依赖于KRAS和自噬,但是KRAS在自噬方面的作用仍然未知,这篇研究表明KRAS的抑制和ERK MAPK的药理抑制增加了自噬通量,而且削弱了糖酵解和线粒体功能。作者设想抑制ERK可以增强PDAC对自噬的依赖,这可能是通过损害KRAS或ERK驱动的代谢过程。他们发现自噬抑制剂和特异性自噬调节因子的遗传或者药理学抑制,协同增强了ERK抑制剂介导KRAS驱动的PDAC中抗肿瘤的能力。最终结论:ERK MAPK(或者ERK)和自噬过程的药物抑制剂组合可能是治疗PDAC的有效方法。
KRAS突变是胰腺癌(PDAC)发生发展的关键遗传驱动,并且是维持PDAC肿瘤生长的必要条件。鉴于95%的PDAC都有KRAS驱动突变,美国国家癌症研究所已经将抗KRAS疗法确定为胰腺癌研究的4个优先事项之一。KRAS突变的PDAC的特点是发生多种代谢改变,包括糖酵解、谷氨酰胺降解、自噬依赖性增加。现在KRAS抗体药物至少有5个主要的方向。很有前景的方案是针对调节KRAS依赖代谢功能,这些功能支持PDAC发生发展的能量需求。其中一个是自噬和噬菌体调节的过程,即细胞降解细胞器、大分子和细胞废物循环利用,由此产生的分解产物作为生物能中间体,以维持代谢需求。因为自噬在KRAS突变的PDAC中上调,并且对肿瘤生长很重要,所以自噬抑制剂羟氯喹正在接受临床评估。羟氯喹作为单药治疗是很有限的,但是和紫杉醇联合治疗很有前景。这篇研究提供了一个新的治疗策略:RAF-MEK-ERK级联抑制剂破坏KRAS驱动的代谢过程,促使PDAC严重依赖自噬,从而增强羟基氯喹对自噬的抑制性。
KRAS抑制提高自噬通量在KRAS突变的人胰腺癌细胞系中,基本自噬水平非常高。为了确定KRA S突变对维持高水平的自噬是否是必须的,作者利用3种技术来研究KRAS急性抑制对自噬通量的影响。作者首先评估了稳定表达串联荧光报告基因mCherry-EGFP-LC3B的PDAC细胞株的自噬通量。LC3B是自噬相关蛋白,它经过翻译后修饰,使其脂化并和自噬泡相关联。利用之前研究验证过的寡聚siRNA来对KRAS急性抑制,发现6个细胞系的自噬通量增加了2到10倍(图1a,1b)。RAS抑制剂ARS-1620处理KRAS G12C突变的MIA PaCa细胞后,自噬通量也相应增加(图1c)。第二,研究者通过体外表达EGFP-LC3b和体外检测自噬体相关点状形成来检测自噬体组装。结果表明抑制KRAS时观察到的点状增加是由于自噬通量增加,而不仅仅是稳态变化所致。第三,研究者通过免疫印迹技术来检测内源性LC3B-Ⅰ向自噬体相关的脂化LC3B-Ⅱ的转化。siRNA引起的KRAS抑制增加了LC3B-Ⅱ/LC3B-Ⅰ的比率,并且当bafilomycin A1抑制自噬体降解,这增加的比例是不变的(图1d)。因此,在KRAS突变的PDAC细胞系中,抑制KRAS增加了自噬通量,同时自噬体生成、溶酶体融合和自噬体相关LCB-Ⅱ也提高了。
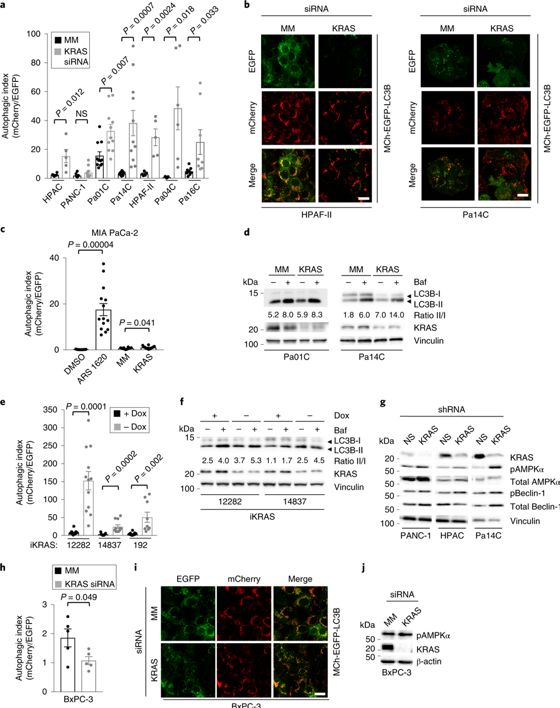
图1
作者接下来在Kras驱动的PDAC小鼠模型(iKRAS)中进行研究,通过模型建立的三个肿瘤细胞系中,在用药物处理后Kras G12D被抑制(24h),自噬通量增加了7-20倍(图1e),并且LC3B-Ⅱ/LC3B-Ⅰ的比例也同样增加(图1f)。因此在人、鼠的PDAC中,突变KRAS的抑制导致自噬增加而非降低。
AMPK和Beclin-1是早期自噬形成和噬菌体组装的关键驱动因子。已有研究表明KRAS沉默诱导自噬,研究者接下来测试KRAS变化是否影响上游自噬通路。他们发现shRNA介导KRAS的抑制增加了KRAS突变的PDAC细胞系和iKRAS细胞系中AMPK磷酸化,并且激活或增加了总Beclin-1的水平(图1g)。另外,将10个KRAS突变PDAC细胞系进行RNA-seq,并对测序数据进行基因集变异分析(GSVA)发现在9个细胞系中短暂的shRNA抑制KRAS和编码自噬蛋白的基因转录增加有关。KRAS突变的PDAC细胞系中观察到的自噬通量和AMPK活化增加,而在KRAS野生型PDAC细胞系BxPC-3中,抑制KRAS这两种情况都没有发生(图1h,1j)。在两种不同的胰腺细胞模型中,得出相同结论:KRAS可上调自噬,这是PDAC的关键生存机制。
ERK抑制提高自噬通量作者发现ERK1/ERK2丝裂原活化蛋白激酶对KARS突变的PDAC的体内、体外生长有重要作用。为了确定这个关键KRAS效应通路是否对抑制KRAS诱导的自噬同样重要,作者用ERK1/ERK2选择性抑制剂SCH772984(ERKi)处理8个PDAC细胞系(稳定表达mCherry-EGFP-LC3B自噬报告基因)24小时,发现自噬通量增加了3-30倍(图2a,2b)。ERKi处理的细胞和对照相比,LC3BⅡ/LC3BⅠ比率增加(图2c),这也证实自噬通量增加。在iKRAS小鼠模型中,作者观察到ERK抑制同样使3个小鼠iKRAS PDAC细胞系的Kras G12D沉默,这导致自噬通量大幅度增加10-30倍(图2d,2e)。
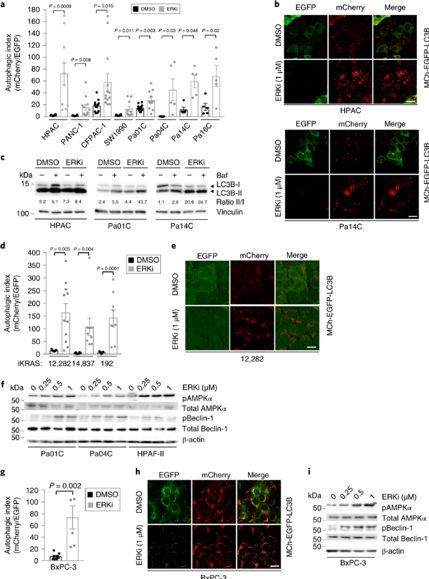
图2
作者检测了ERKi处理后AMPK和Beclin-1的活化,发现在抑制KRAS时,抑制ERK也导致人PDAC细胞中AMPK和Beclin-1磷酸化增加(图2f)。尽管在某些细胞系中PRKAA2和BECN1转录增强,但ERKi处理1小时内磷酸化的Beclin-1水平也是增加的,这表明在PDAC中KRAS通过ERK MAPK级联反应调节自噬。BxPC-3细胞系尽管缺乏KRAS突变,但仍有较高的自噬能力,这是由于ERK信号上调以及ERK依赖下游突变BRAF生长所致。和在调节自噬中ERK信号的关键作用一致的是,BxPC-3细胞系经过ERKi处理后同样表现出自噬通量增加。
抑制ERK可以减少代谢活动作者对ERKi诱导的自噬通量增加的基础机制进行了研究。首先对12个PDAC细胞系用ERKi或者DMSO处理1小时和24小时,然后进行反相蛋白微阵(RPPA)分析(图3a)。比较所有蛋白的均值后发现:ERK抑制的有效标记物减少,包括ERK1/ERK2的磷酸化和活化、ERK底物RSK的磷酸化、ERK调节底物MYC总蛋白的减少。ERKi处理24小时后,凋亡介质BIM、PUMA和caspase 7等都被上调。作者进一步验证了抑制ERK可导致AMPKK的活化,另外还观察到mTORC1信号的减弱(图3a,3b)。这两个信号通路在ERKi刺激下的改变是促进自噬的主要机制。RPPA数据的相关性分析表明磷酸化的AMPK水平和ERK抑制标记物呈负相关,而mTOR通路中成分降低和相同标志物呈正相关。

图3
作者接下来利用LC-MS/MS对一组经ERKi处理的PDAC细胞系进行极性代谢物水平分析。和RPPA所确定的AMPK活性增加相一致,代谢组学分析也表明ERKi处理的PDAC细胞系中AMP水平有所增加,并且AMP前体IMP和AMP降解产物也有所增加(图3c)这表明ERKi通过改变核苷酸代谢来增加自噬通量。此外,作者对7株ERK刺激超过24小时的人PDAC细胞系进行了RNA-seq分析,并利用GSVA技术,确定ERKi介导的自噬相关基因和溶酶体基因的表达增加(图3d,3e)。
RNA-seq分析发现自噬的基因转录上调,但诱导ERK抑制的糖酵解基因转录本减少(图3f)。用葡萄糖LC-MS和NMR进行代谢通量分析来验证这些变化导致糖酵解活性下降。在用[1,6-13C2]孵育细胞之前,作者用siRNA处理细胞,使KRAS基因沉默,发现KRAS抑制和ERK抑制降低了葡萄糖摄取。因此在ERKi处理的7株KRAS突变PDAC细胞中,关键糖酵解中间体明显减少。iKRAS PDAC小鼠模型中的分析表明,突变激活的KRAS驱动糖酵解活性,而Kras G12D的抑制降低了它的活性。作者认为突变激活的KRAS调节了人PDAC细胞ERK抑制后有效抑制的糖酵解活性。作者假设这种ERK抑制诱导的糖酵解活性的降低有助于ERK抑制剂介导的自噬能通量的增加。当去除PDAC细胞的生长培养基中的葡萄糖时,自噬通量增加,但幅度小于ERK抑制。因此,ERK抑制剂治疗可直接和间接减少糖酵解来增加自噬通量。
ERK抑制破坏线粒体活性RNA-seq数据显示ERKi处理导致线粒体相关基因转录水平降低(图3g)。作者之前的研究表明能长期存活的小鼠PDAC细胞系罕见亚型(<10%)更依赖于线粒体呼吸。因此,KRAS沉默或者抑制ERK降低了线粒体相关基因的转录,这是出人意料的。作者首先评估了短期KRAS抑制和ERK抑制对线粒体形态和活性的影响,ERKi处理减少了线粒体数量(图4a)。抑制ERK使有丝分裂通量增加,总PINK1水平增加。因此线粒体成分的转录减少,增加的有丝分裂弥补了线粒体数量的减少。作者发现ERK抑制剂处理PDAC细胞,1小时内诱导了线粒体融合(图4b,4c),这和DRP1(线粒体分裂中间物和ERK底物)的磷酸化水平降低有关(图4d)。ERKi处理24小时候线粒体融合、DRP1磷酸化水平降低(图4e,4f)。同时,DEP1总蛋白含量下降(图4g),这和线粒体相关基因转录水平下降是一致的。线粒体在ERK抑制下融合增加,这和人PDAC细胞系中KRAS短期抑制、在iKRAS小鼠模型中Kras G12D抑制的现象一致。总的来说,ERK抑制和KRAS抑制都会导致线粒体网络的明显重排。

图4
发现在人PDAC细胞系和iKRAS小鼠模型中线粒体融合是和线粒体活性增加有关(图4i,4j)。在ERKi处理1.5小时后细胞耗氧率并没有变化(图4k),而在这个时间段线粒体融合是很强的。因此当人PDAC细胞用ERKi处理24小时,基础耗氧率和ATP产生出现降低或者不变的现象(图4l)。在人PDAC细胞抑制KRAS、在iKRAS小鼠模型关闭Kra G12D的表达后都出现相似的现象。在Kras抑制的小鼠模型以及ERKi抑制的PDAC细胞系中备用线粒体含量小幅增加(图4l),这和线粒体电位增加相一致。由于作者并没有观察到耗氧量和ATP的变化,因此这增加的电位并不是用来产生能量。
抑制ERK增强了PDAC对自噬的依赖性作者假设在抑制ERK时观察到的自噬增加可能是PDAC细胞对自噬产生更强的依赖性。若如此,那ERK抑制和自噬同时发生应该比单独发生更加有效。临床上没有特异的自噬抑制剂,因此利用羟氯喹-溶酶体酸化的常用抑制剂-来作为自噬的间接抑制剂。作者发现用SCH772984(ERKi),或MEK抑制剂(MEKi)和羟氯喹同时处理可增强生长抑制(图5a,5b)。利用Chou-Talalay或者BLISS方法观察到ERKi或MEKi的协同作用(图5c)。另外,联合处理引发的自噬量比ERKi单独处理增加的多(图5d)。而且当在人胰腺受试者衍生的器官样培养物上评估时,ERKi和羟氯喹之间的协同作用是不变的。(图5e,5f)。

图5
为了将这些体外发现在肿瘤内验证,作者在两个PDX小鼠模型中评估了ERKi和羟氯喹联合作用。在两个模型中ERKi单独处理对肿瘤生长有一定的影响,但联合处理抑制了肿瘤发生并且生存率有所提高(图5g)。另外ERKi和羟氯喹同时处理小鼠,肿瘤大小明显比ERKi单独处理的小(图5h)。因此ERK1/ERK2和自噬的联合抑制对抑制PDAC生长更加有效。
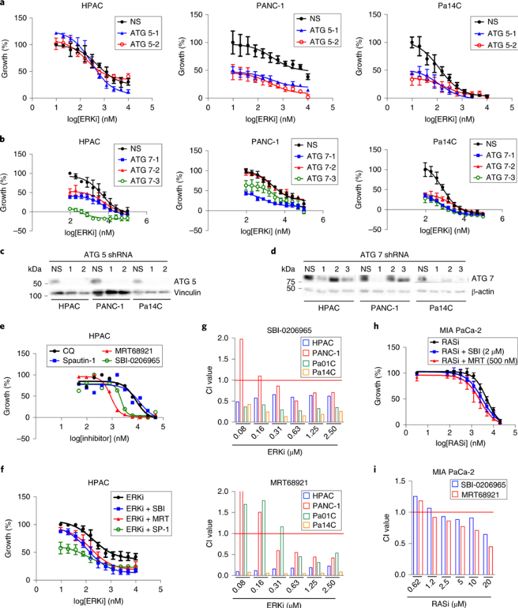
图6
因为羟氯喹间接抑制自噬体,作者接下来确定自噬的特定组分的遗传或药理学阻断是否也与抑制ERK发生协同作用。作者首先利用shRNA抑制自噬相关基因(ATG5和ATG7),发现在PDAC细胞系中,ATG5或ATG7抑制显著增强了ERKi引发的细胞生长抑制(图6a,6d)。现在已开发出更加特异的自噬抑制化合物,包括SBI-0206965(SBI),MRT68921(MRT)和Spautin-1。SBI靶向作用ULK1,MRT靶向作用ULK1和ULK2(AMPK磷酸化的自噬预启动复合物的组分)。Spautin-1抑制两种特异性肽酶USP10和USP13,它们调节Vps34复合物中Beclin-1的泛素化,从而启动自噬体形成。 与羟氯喹一样,这三种自噬抑制剂都能减少PDAC细胞系的增殖(图6e)。ATG5和ATG7基因抑制后,和三种药理抑制剂联合处理增强了ERK I介导的生长抑制(图6f,6g)。由于KRAS G12C突变体MIA-PaCa-2细胞对氯喹具有抗性,作者用ARS-1620(RASi)与SBI或MRT组合处理它们,发现ARS-1620诱导的生长抑制的协同增强以及联合处理的细胞中有显着增强的细胞凋亡(图6h,6i)。
本网站所有内容来源注明为“williamhill asia 医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于williamhill asia 医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“williamhill asia 医学”。其它来源的文章系转载文章,或“williamhill asia 号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与williamhill asia 联系,williamhill asia 将立即进行删除处理。
在此留言






#Nat#
50
#ERK#
57
#Med#
58