
Nature:突破欧洲血统局限:MAGE数据集解锁多种族基因表达奥秘
13小时前 生物探索 生物探索 发表于上海

MAGE项目强调了在遗传研究中纳入多样化样本的重要性。这不仅提高了因果变异识别的分辨率,还为研究这些变异在不同人群中的作用机制提供了更全面的视角。
引言
遗传变异影响基因表达和剪接,是导致人类表型多样性的主要因素之一。尽管这类研究非常宝贵,但其参与者主要是欧洲血统人群,导致结果的普适性受限,并且对进化研究的贡献有限。为了解决这些问题,7月17日Nature的研究报道“Sources of gene expression variation in a globally diverse human cohort”,开发了MAGE(Multi-Ancestry Gene Expression),一个开放获取的RNA测序数据集,涵盖了来自1000 Genomes Project的731名个体,分布于全球五大洲的26个种群。
多年来,基因表达与剪接的遗传变异研究为揭示这些分子特征与更高层次表型之间的关系奠定了基础。然而,这些研究往往忽略了非欧洲血统的个体,限制了研究结果的广泛应用性。例如,遗传关联研究(GWAS)大多集中在欧洲血统人群,这可能导致在其他种群中的预测准确性降低,并且难以发现那些在其他种群中特有的重要变异。因此,研究多样化人群中的基因表达和剪接变异显得尤为重要。
MAGE项目通过对1000 Genomes Project中731个淋巴母细胞系的RNA测序,量化了基因表达和剪接的多样性,提供了高分辨率的基因变异与基因表达和剪接之间的关联图(cis-eQTL和cis-sQTL)。研究发现,大部分基因表达(92%)和剪接(95%)的变异存在于种群内部,这与DNA序列变异的分布情况一致。研究人员还发现了超过15000个潜在的因果eQTL和超过16000个潜在的因果sQTL,其中有1310个eQTL和1657个sQTL主要存在于未被充分研究的种群中。
这些发现表明,不同种群间基因表达和剪接的效应大小和方向高度一致,之前观察到的“种群特异性”效应主要是由于研究分辨率低或未检测到的独立eQTL所致。这意味着通过更高分辨率和更全面的数据集,williamhill asia 可以更准确地理解基因表达和剪接的遗传变异。
MAGE项目不仅扩展了williamhill asia 对人类基因表达多样性的理解,还为研究人类基因组的进化和功能提供了一个包容性资源。通过结合现有的全基因组测序数据,MAGE将成为未来研究人类基因表达和剪接多样性的宝贵资源,推动个性化医疗和进化生物学的研究进展。
总的来说,MAGE项目强调了在遗传研究中纳入多样化样本的重要性。这不仅提高了因果变异识别的分辨率,还为研究这些变异在不同人群中的作用机制提供了更全面的视角。通过MAGE,能够更好地理解基因表达和剪接变异对人类表型多样性的贡献,从而为个性化医疗提供更有力的科学依据,并推进基因组学领域的发展。

在人类基因组学研究中,基因表达和剪接的遗传变异是影响表型多样性的重要因素。然而,现有的大多数研究主要集中在欧洲血统的个体,这限制了研究结果的普适性,并且在很大程度上妨碍了对不同人种之间遗传差异的理解。为了填补这一空白,研究团队开发了MAGE(Multi-Ancestry Gene Expression),一个开放获取的RNA测序数据集,涵盖了来自1000 Genomes Project的731名个体,这些个体分布在全球五大洲的26个种群中。
MAGE项目通过对1000 Genomes Project中731个淋巴母细胞系(LCLs)的RNA测序,量化了基因表达和剪接的多样性。研究样本分布在五大洲的26个种群中,每个种群包含27-30个个体,确保了数据的多样性和代表性。
所有样本均在一个实验室内通过17个批次进行测序,样本群体在批次之间进行分层,以避免群体和批次之间的混杂。基因表达水平使用GENCODE (v.38)的基因注释进行量化,替代剪接模式则采用了LeafCutter方法进行量化。
为了确保数据的准确性和可靠性,研究人员使用了一系列生物信息学工具和软件,包括:
ADMIXTURE:用于分析遗传混合情况。
Plink:进行基因型数据处理和质量控制。
Salmon和tximport:用于转录本定量。
STAR:进行RNA序列比对。
LeafCutter:用于量化和分析替代剪接事件。
regtools:结合RNA-seq数据和变异信息进行分析。
MANTA:用于结构变异检测。
DESeq2:进行差异表达分析。
此外,研究还使用了线性模型来评估不同种群间的基因表达和剪接变异,控制了测序批次和性别等因素对结果的影响。
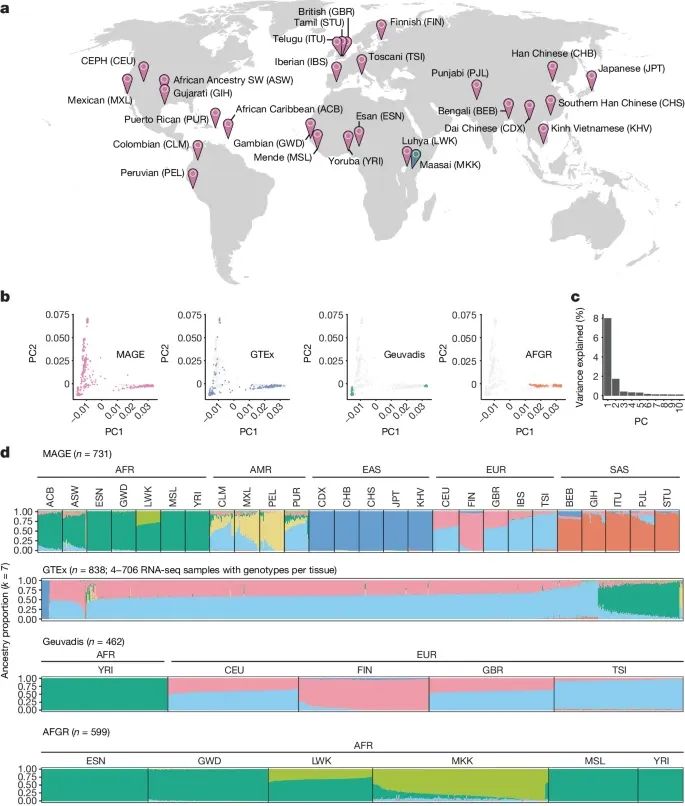
MAGE数据集中基因表达和剪接变异的全球多样性(Credit: Nature)
RNA测序数据的来源:图a显示了来自1000 Genomes Project(1KGP)的731个个体的淋巴母细胞系(LCLs)的RNA测序数据,这些个体分布在全球五大洲的26个种群中。每个种群大约有27-30个个体。
图b-d展示了不同种群和大洲组之间的基因型主成分分析(PCA)和ADMIXTURE分析的结果,这些结果显示了不同种群之间的遗传差异和混合情况。
基因型主成分分析(PCA):图b显示了MAGE数据集与其他大型RNA和全基因组测序数据集(如Geuvadis、GTEx和AFGR)的基因型主成分1(PC1)和主成分2(PC2)的比较。结果表明,MAGE数据集包括了以前研究中未充分代表的非洲血统和其他非欧洲血统的群体。图c展示了前十个主成分解释的变异比例,显示了基因型数据在不同种群之间的变异情况。
ADMIXTURE分析:图d显示了不同个体基因组成分的ADMIXTURE分析结果。结果表明,不同个体的基因组成分可以归因于不同的祖先成分,这些成分在不同种群中有显著的差异。
基因表达和剪接多样性
研究发现,基因表达(92%)和剪接(95%)的变异主要分布在种群内部,而不是种群之间,这与DNA序列变异的分布情况一致。这一发现表明,尽管不同种群之间存在遗传差异,但大多数基因表达和剪接的变异实际上是在个体内部进行的。
遗传变异对基因表达的影响
通过MAGE数据集,研究人员绘制了遗传变异与邻近基因的表达和剪接(分别称为cis-eQTL和cis-sQTL)之间的关联图,发现了超过15000个潜在的因果eQTL和超过16000个潜在的因果sQTL。这些QTLs富集在相关的表观基因组特征中,表明它们在基因调控中具有重要作用。
特定种群中的QTL
其中有1310个eQTL和1657个sQTL主要存在于未被充分代表的种群中。例如,在非洲裔、拉丁美裔等群体中,研究识别出一些特有的QTLs,这些QTLs在欧洲血统的个体中很少甚至不存在。这些发现对于理解不同人群中的遗传变异具有重要意义。
QTL效应的一致性
研究表明,因果eQTL效应的大小和方向在不同种群中高度一致。以往研究中观察到的“种群特异性”效应主要是由于分辨率低或未检测到的同一基因的其他独立eQTL所致。这一发现对预测应用(如多基因风险评分和转录组关联研究)具有重要意义,因为它表明,如果能够充分控制全局遗传背景的影响,因果变异的效应在不同种群之间通常不会有显著差异。
进化选择的证据
研究还揭示了选择压力对基因表达变异的影响。通过对选择约束指标(如pLI (probability of loss-of-function intolerance))的分析,发现与基因表达相关的变异在多个细胞和组织类型的调控区域中高度富集。这表明,自然选择在塑造人类基因表达变异方面发挥了重要作用。
功能注释和共定位分析
研究人员还进行了功能富集分析和共定位分析,以探索基因表达和剪接变异的功能意义和潜在机制。结果表明,许多eQTL和sQTL与已知的调控区域和表观基因组特征高度重叠,这进一步支持了这些变异在基因调控中的重要作用。例如,在调控基因表达的启动子区域和增强子区域发现了显著富集的eQTL信号。
与复杂性状的关联
为了进一步探讨基因表达相关的遗传变异在复杂性状中的作用,研究团队还分析了这些QTLs与已知的全基因组关联研究(GWAS)信号的共定位情况。结果发现,有54个GWAS信号与MAGE中的eQTL和sQTL信号共定位,这表明这些QTLs可能在调控这些复杂性状中发挥重要作用。例如,一个与血小板计数相关的GWAS信号与TPM4基因的eQTL共定位,这提示该基因可能在血小板功能中具有重要作用。
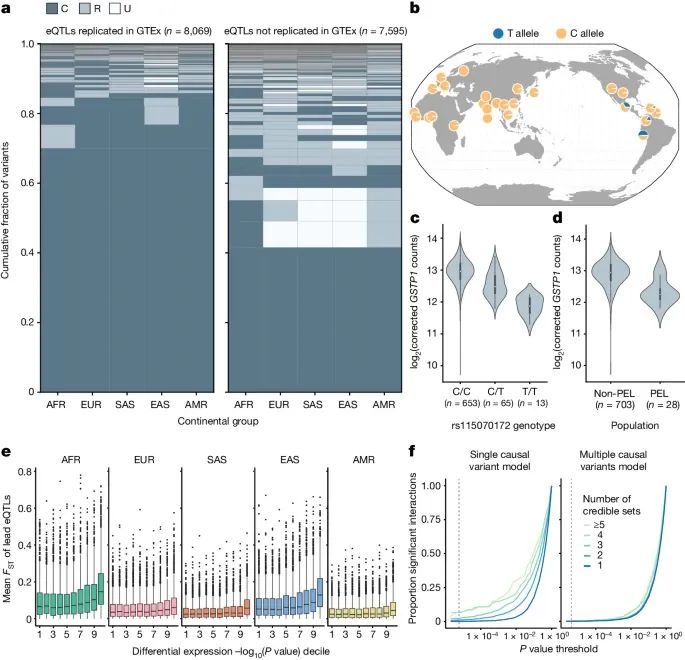
不同种群中的eQTL频率和效应存在显著差异,这些差异对基因表达水平有重要影响(Credit: Nature)
eQTL的全球频率分布:图a展示了MAGE数据集中15,664个细分eQTL在不同大洲群体中的等位基因频率(AF)分布。结果表明,不同种群中的eQTL等位基因频率差异显著,部分eQTL在某些种群中特有或更为常见。
特定种群的eQTL示例:图b和图c详细展示了一个特定eQTL(rs115070172)的等位基因频率和基因表达关联。这个eQTL在混合美洲群体中T等位基因频率较高(AF > 0.05),在秘鲁人群中频率更高(AF = 0.63)。T等位基因显著关联于GSTP1基因表达水平较低。
不同群体间的基因表达差异:图d显示了GSTP1基因在秘鲁人群(PEL)和其他全球人群中的表达水平差异,结果表明秘鲁人群中的GSTP1基因表达显著低于其他群体。这种差异可能是由特定eQTL的频率分布差异引起的。
eQTL与基因表达差异的关联:图e展示了各大洲群体之间的基因表达差异(FDR调整后的P值≤0.05)与eQTL的FST值之间的关联。结果表明,基因表达差异显著的基因其eQTL具有较高的FST值,表明eQTL频率差异在不同种群间基因表达差异中起重要作用。
eQTL效应大小的一致性:图f比较了单一因果变异模型和多个潜在因果变异模型中显著基因型与大陆群体间相互作用的数量。结果显示,在考虑单一因果变异的模型中,有70个eQTL表现出显著的相互作用效应(Bonferroni校正后)。然而,当考虑多个潜在因果变异时,这些相互作用效应大多消失,表明多因果变异的加性效应可能导致了表观的相互作用效应。
MAGE项目的开发和分析不仅扩展了williamhill asia 对人类基因表达和剪接多样性的理解,还为研究人类基因组的进化和功能提供了一个包容性资源。通过结合现有的全基因组测序数据,MAGE将成为未来研究人类基因表达和剪接多样性的宝贵资源,推动个性化医疗和进化生物学的研究进展。
这项研究还强调了在遗传关联研究中纳入多样化样本的重要性。通过增加样本的多样性,可以提高因果变异识别的分辨率,并更全面地理解这些变异在不同人群中的作用机制。总的来说,MAGE项目为理解基因表达和剪接变异对人类表型多样性的贡献提供了重要的科学依据,为未来的基因组学研究提供了重要的参考和数据支持。
MAGE数据集的开发和应用展示了在多种族背景下研究基因表达和剪接变异的巨大潜力。通过高分辨率的QTL绘图和丰富的功能注释,这项研究为未来的基因组学和个性化医学研究提供了宝贵的资源和新的见解。
参考文献
Taylor DJ, Chhetri SB, Tassia MG, Biddanda A, Yan SM, Wojcik GL, Battle A, McCoy RC. Sources of gene expression variation in a globally diverse human cohort. Nature. 2024 Jul 17. doi: 10.1038/s41586-024-07708-2. Epub ahead of print. PMID: 39020179.
https://www.nature.com/articles/s41586-024-07708-2
本网站所有内容来源注明为“williamhill asia 医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于williamhill asia 医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“williamhill asia 医学”。其它来源的文章系转载文章,或“williamhill asia 号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与williamhill asia 联系,williamhill asia 将立即进行删除处理。
在此留言













前往app查看评论内容
1 0