
读书报告 | 阻断基因组不稳定性预防黑色素瘤对MAPK抑制剂获得性耐药
2023-12-27 iCombo iCombo 发表于上海

本文提供给williamhill asia 一个关键的治疗思路,即靶向基因组不稳定性的起因能够预防获得性耐药。
导读
阻断癌症基因组不稳定性可能会阻止肿瘤多样化和治疗耐药。本文发现,在患者和患者来源的异种移植物模型(PDXs)中使用MAPK抑制剂(MAPKi)治疗后,转移性皮肤黑色素瘤的获得性耐药基因组通过复杂基因组重排(CGRs)和染色体外DNA(ecDNAs)特异性扩增耐药驱动基因、非同源末端连接(NHEJ)和同源重组修复(HRR)基因。几乎所有敏感和获得耐药基因组都有普遍存在的泛发性嗜色区(pervasive chromothriptic regions),具有不成比例的高突变负荷,并且与ecDNA和CGR跨度有显著重叠。ecDNA和CGR扩增子内的体细胞突变经常富集HRR标签,特别是在获得性耐药肿瘤内。无论敏感还是耐药,断裂点序列分析均表明,NHEJ对于CGR和ecDNA形成的双链DNA断裂修复至关重要。在人类黑色素瘤细胞系和PDXs中DNA-PKCS抑制剂靶向NHEJ可减少ecDNAs和CGRs和预防/延迟获得性MAPKi耐药。因此,本文提供给williamhill asia 一个关键的治疗思路,即靶向基因组不稳定性的起因能够预防获得性耐药(Cancer Discov. 2023 Apr 3;13(4):880-909. doi: 10.1158/ 2159-8290.CD-22-0787)。
Introduction
逆转耐药进化的科学努力主要集中在靶向获得性弱点。相比之下,在接受靶向治疗的患者中,导致癌症快速发生耐药的机制(例如基因组不稳定通路)仍知之甚少。
患者对BRAFi + MEKi治疗的获得性耐药是通过高频基因扩增发生的,单独或联合基因扩增可重新激活MAPK通路。
皮肤黑色素瘤是一种在预先未接受过靶向治疗的情况下,染色体碎裂负担本底很高的癌症,而染色体碎裂似乎是癌症快速产生和积累高度动态结构变异体(SVs)的关键进化机制。与SV相关的扩增子可以被识别为染色体内复杂基因组重排(CGRs)和染色体外DNA(ecDNAs,又名double minutes),它们可能是时间相关的结构,赋予一系列基因组信号可塑性。ecDNAs的非孟德尔遗传、其可及的染色质,以及ecDNAs劫持增强子和转录枢纽聚集,都是促进快速适应极端应激(如癌基因靶向治疗)的机制。
晚期皮肤黑色素瘤队列和全基因组测序数据特征
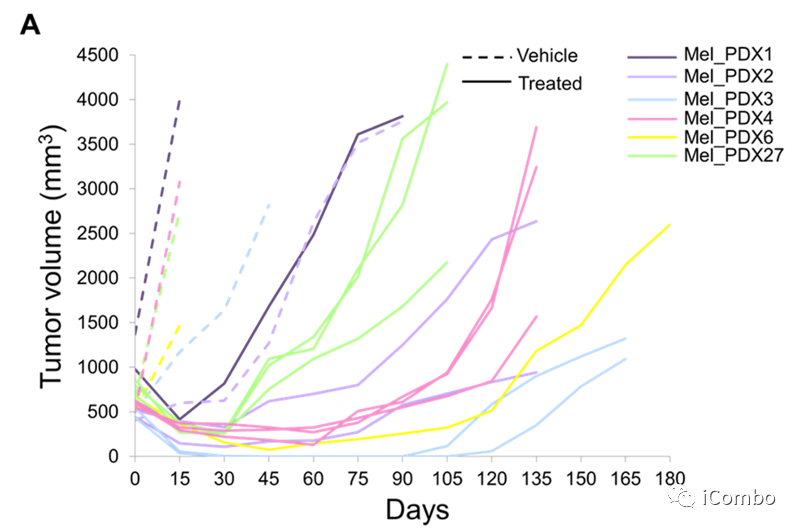
第一个队列包括患者匹配的正常组织以及MAPKi治疗前的BRAFV600MUT黑色素瘤,以及初始缓解后的疾病进展时的组织(n=10个正常组织;n=10个治疗前肿瘤;n=17例获得性耐药肿瘤;N=10位病人)。第二组由快速尸检的黑色素瘤组织构成(n=3个正常组织;1例为BRAFV600MUT敏感肿瘤;获得性耐药肿瘤12例;n=3例死亡受试者,均接受MAPKi治疗;n=6个转移器官部位)。第三个队列由BRAFV600MUT或NRASMUT PDX肿瘤组成。本研究对PDXs(N=6;在NOD-scid IL2R gamma null(NSG)小鼠中,1个BRAFV600MUT模型和5个NRASMUT 模型)的MAPKi治疗的剂量足以引起肿瘤消退,然后产生获得性MAPKi耐药肿瘤(n=6个对照治疗的肿瘤;获得性耐药肿瘤12例;N=6例正常组织)
获得性耐药 vs. MAPKi-Naïve的黑色素瘤中ecDNA和CGRs的复发、数量、染色体起源和复合体

共58例BRAFV600MUT和NRASMUT的MAPKi敏感/naïve和获得性耐药肿瘤(n=17例敏感肿瘤;N = 41例获得性耐药肿瘤;n=19例病人)
将所有三个队列合并后,williamhill asia 观察到,与敏感相比,获得性耐药的基因组中的ecDNA和CGRs数量较多(获得性耐药:总N =941,每个肿瘤n=31;敏感:总N=241,平均每个肿瘤16个;配对t-test,P=0.09)
CGR-和ecDNA-扩增子驱动MAPKi耐药
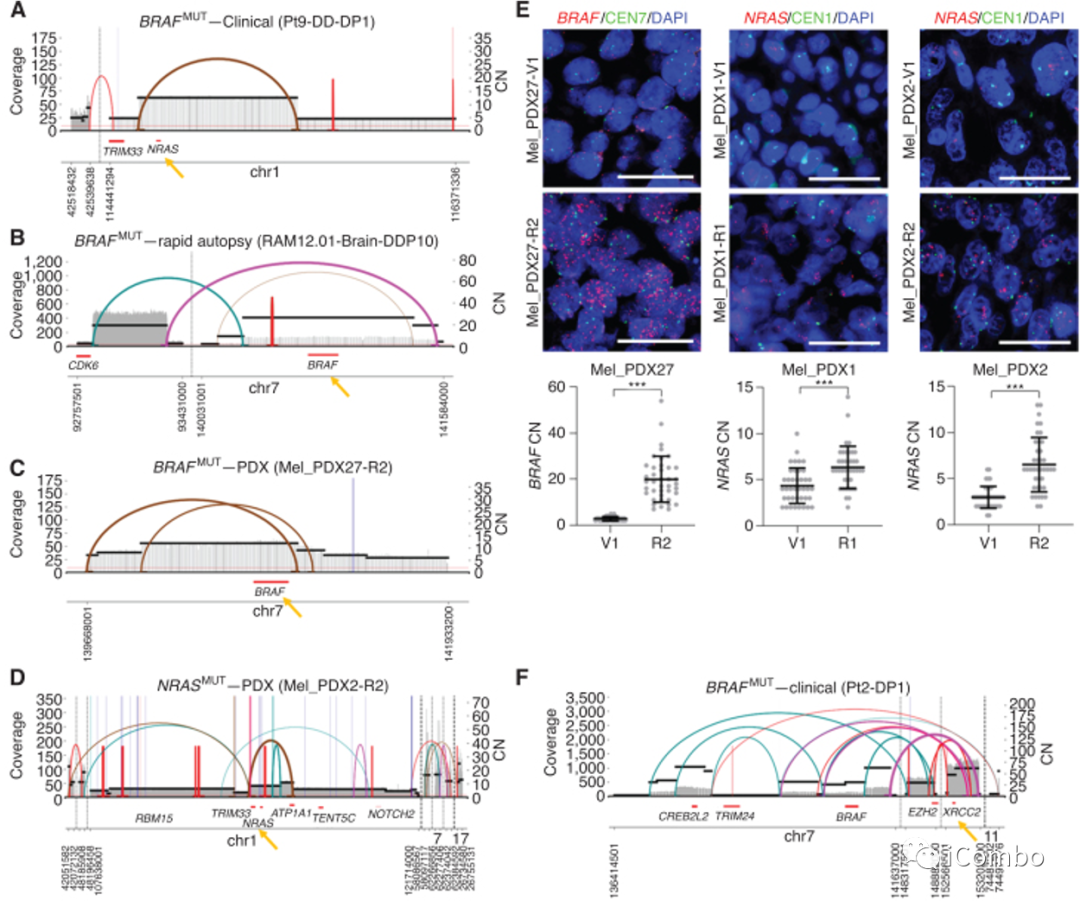
A~D:染色体内CGRs和ecDNA导致的扩增子分析发现获得性耐药肿瘤和携带真正MAPKi耐药驱动基因的CGRs/ ecDNA之间有显著的相关性(P=0.0002)。已知BRAF、NRAS、HRAS、MYC和EGFR,在扩增时,可驱动获得性MAPKi耐药和MAPK通路的再激活
A:使用被称为CRISPR-CATCH的新方法,通过直接分离和高深度测序(NRAS扩增)对复发ecDNA进行验证。这一替代技术证实了这一获得性耐药临床肿瘤样本中890kb驱动ecDNA的环状连接
E:使用DNA-FISH交叉验证耐药特异性存在和BRAF/NRAS扩增子的CNs
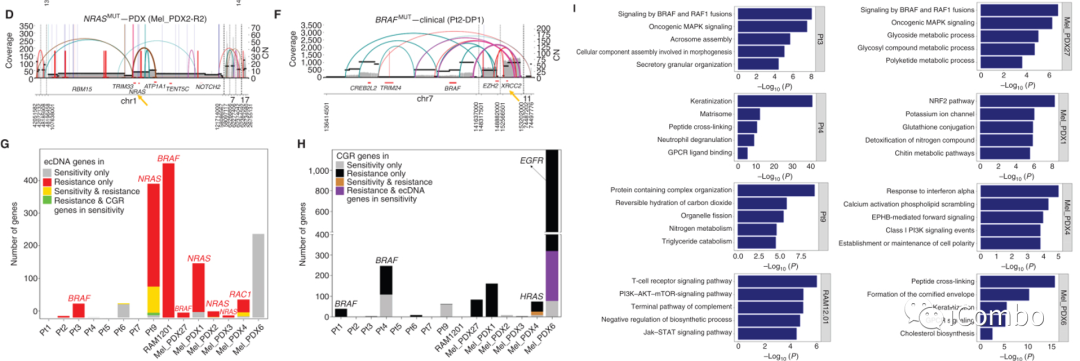
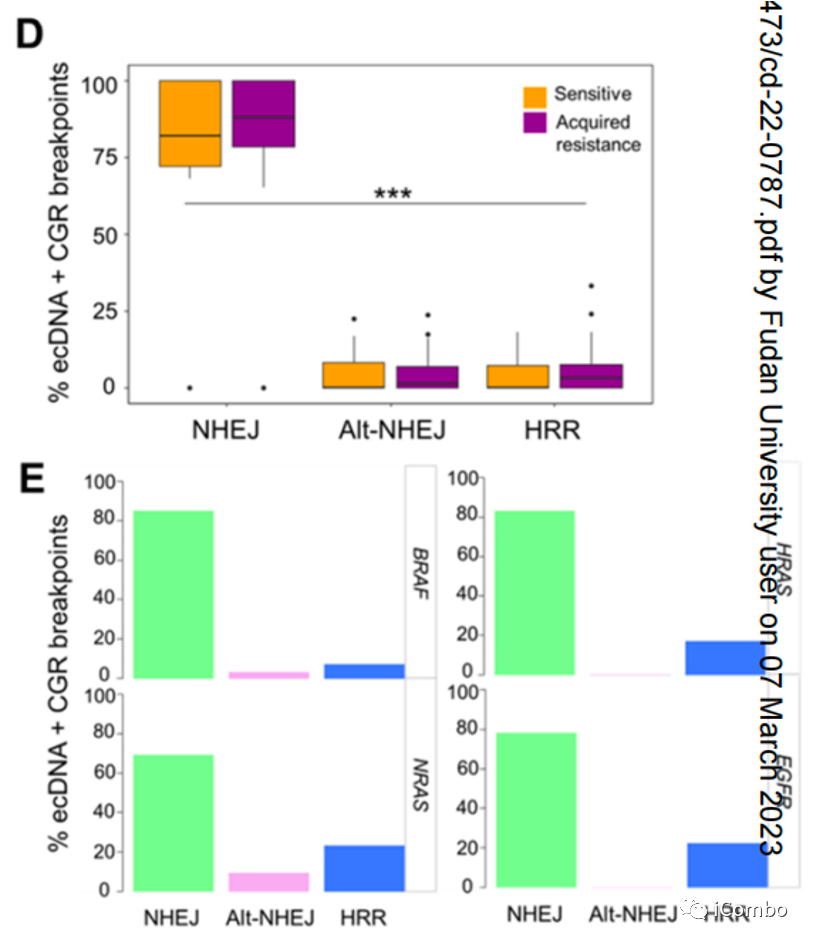
DSBs的NHEJ和HRR被认为是染色体畸变后CGR和ecDNA产生的关键
F:XRCC2(RAD51-like; CN, 49)、一个关键的HRR基因;XRCC6(KU70; CN, 14-15)、一个关键的NHEJ基因;在获得性MAPKi耐药黑色素瘤中特异性扩增为ecDNA
G/H:在ecDNA或CGR扩增子中鉴定出的与敏感、耐药或敏感加耐药特异性相关的耐药特异性基因及其CNs。重要的是,ecDNA和CGRs的基因扩增在MAPKi治疗中促进疾病进展
I:通路富集分析
黑色素瘤的普遍嗜色性基因组跨越与ecDNA和CGRs的基因组坐标重叠
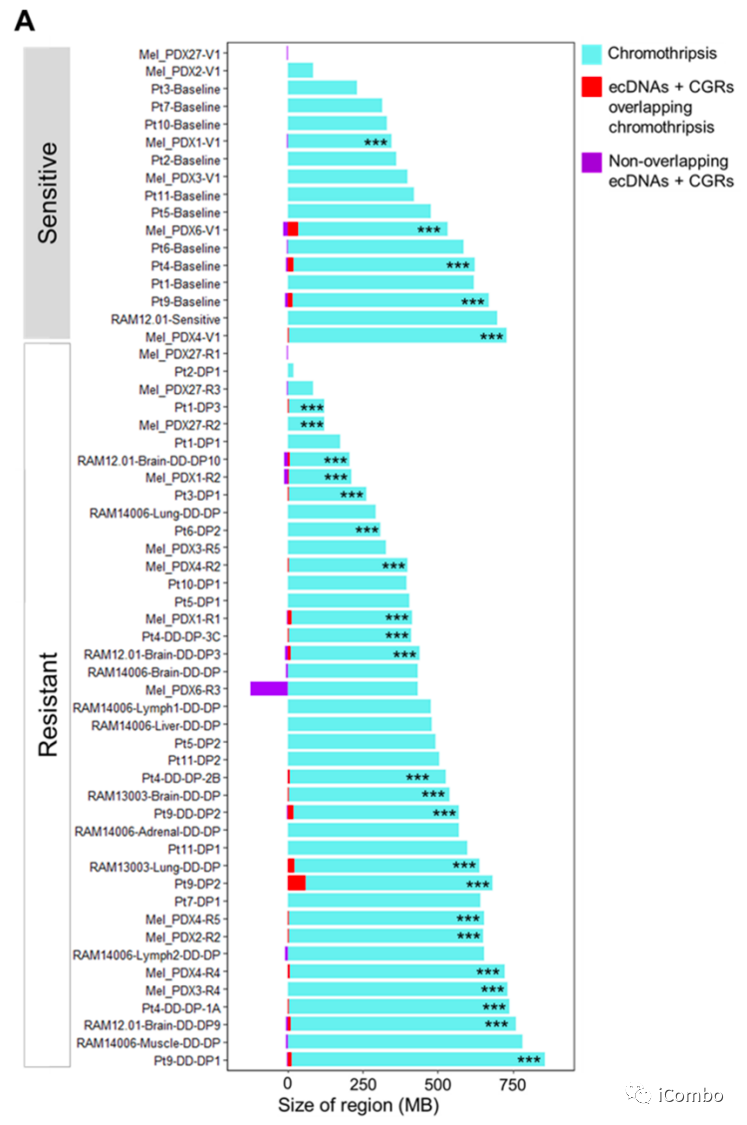
如果染色体碎裂是黑色素瘤疾病进展期间产生ecDNA和CGR的前体步骤,williamhill asia 预计受累基因组跨度会出现非随机重叠
A:在MAPKi敏感/naïve基因组中约29%(5/17)和获得性MAPKi耐药基因组中约~54%(22/41),观察到ecDNAs和CGRs的基因组跨度与显色区显著或非随机重叠
A:在ecDNA+ CGR和染色质区之间有重叠的每个肿瘤基因组中,williamhill asia 观察到显著的非随机收敛(P<0.00001),提示染色体畸变是ecDNA和CGRs的起源
Chromothripsis标签、ecDNAs和CGRs的肿瘤突变负荷(TMBs)和单碱基置换(SBS)
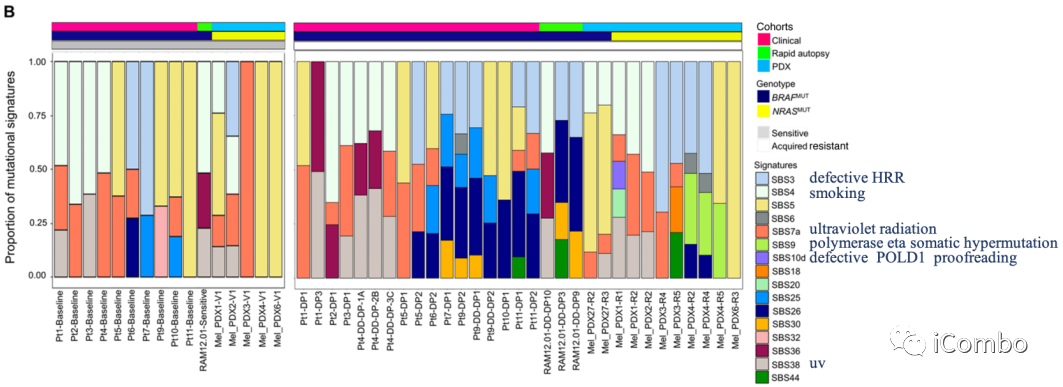
williamhill asia 已经报道了临床黑色素瘤的MAPKi选择改变了突变谱
为确定MAPKi相关的突变体表型如何影响MAPKi选择的染色体稀少基因组,williamhill asia 剔除了MAPKi敏感/naïve(平均,64 SNVs/Mb)vs. 耐药基因组(平均,58 SNVs/Mb)特有的染色体稀少体细胞突变,并分析了SBSs标签
获得性MAPKi耐药的独特和反复发生的是检测到DNA错配修复缺陷(MMR)的突变标签(SBS6, SBS20, SBS26, SBS44;31例耐药肿瘤中14例;16例患者中,10例为基底切除修复(BER)缺损(SBS18、SBS30、SBS36)
在获得性耐药(平均130 SNVs/Mb)肿瘤中,CGR+ ecDNA相关的TMB有高于MAPKi敏感/naïve肿瘤(平均80 SNVs/Mb)的趋势
与患者匹配的敏感肿瘤(1/12)相比,SBS3的富集(缺陷的HRR)在耐药肿瘤的ecDNA/CGRs中更频繁(11/28)
25%(7/28)的获得性耐药肿瘤在ecDNA和CGRs的缺陷HRR标签(SBS3)中显示阳性富集评分,而在敏感肿瘤中仅8%(1/12)
简言之,无论是在MAPKi敏感/naïve还是获得性耐药肿瘤中,ecDNA和CGR扩增子都具有提示HRR特异性缺陷或缺陷的SBS模式
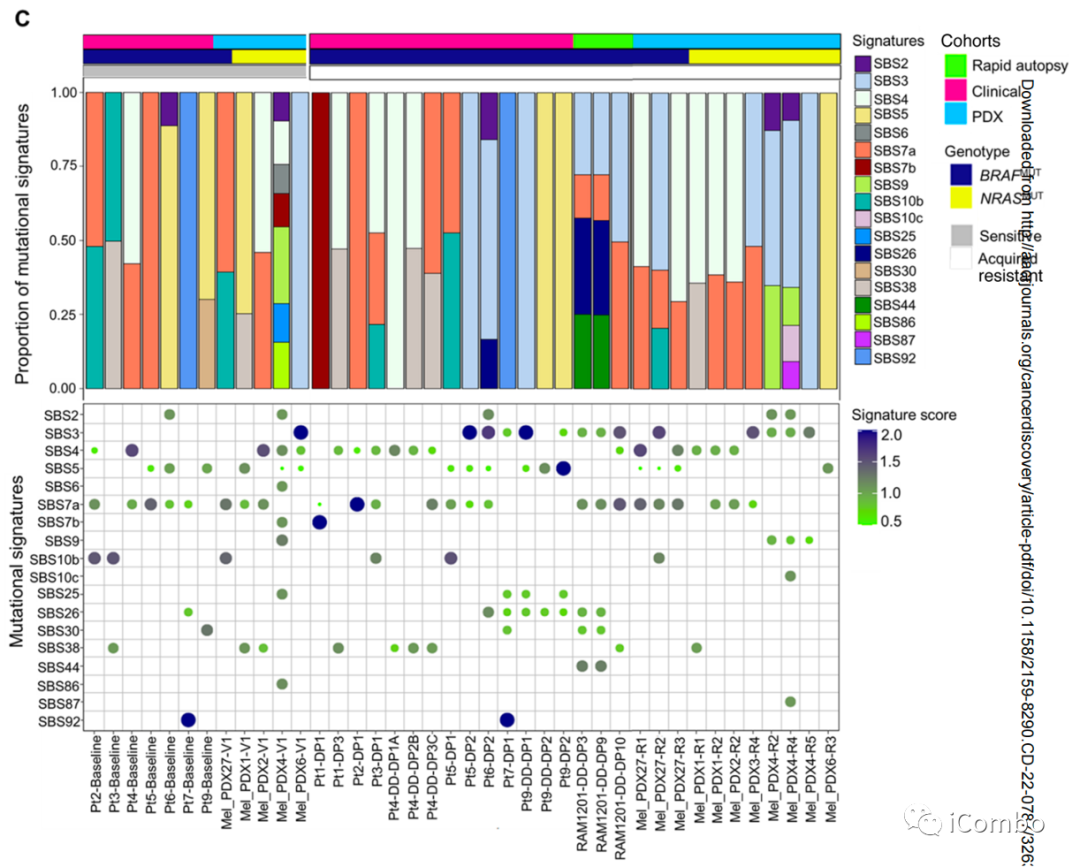
NHEJ是EcDNAs和CGRs形成的基础
D:分析CGRs和ecDNAs的断点连接序列以推断耐药相关扩增子的DSB修复过程。对所有耐药和敏感性相关CGRs和ecDNA的断裂连接序列分析表明,NHEJ是双链DNA片段连接或重排的主要机制。更少的断裂点序列显示短的和长的同源序列以及插入(>10 bp),分别提示通过NHEJ和HRR修复DSB
E:对含有MAPK再激活或MAPKi抗性驱动基因的特异性ecDNAs和CGRs的断点连接序列的分析也揭示了类似的模式,NHEJ在DSBs修复机制中占主导地位
DNA-PKi和/或PARPi预防黑色素瘤细胞系获得性MAPKi耐药
DNA依赖蛋白激酶催化亚基(DNA-PKCS)和PARP1/2参与多种DSB修复通路,尤其是NHEJ(DNA-PKCS)、HRR(DNA-PKCS, PARP1/2)和MMEJ(PARP1/2)
检测了特异性DNA-PKi NU7026和PARP1/2抑制剂ABT888(维利帕利)单独和联合的活性,以阻止人BRAFV600MUT和NRASQ61MUT黑色素瘤细胞株分别用BRAFi+MEKi或MEKi处理中DTPPs克隆的出现
本文进一步假设,一旦慢性MAPKi治疗完全建立,DNA-PKi可以更有效地预防而不是逆转耐药性
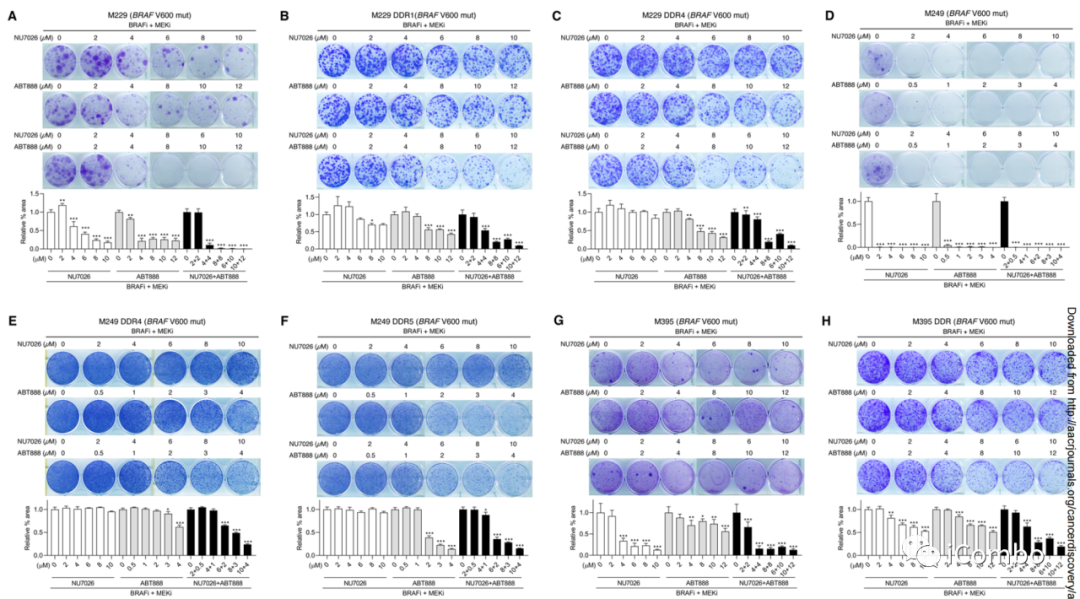
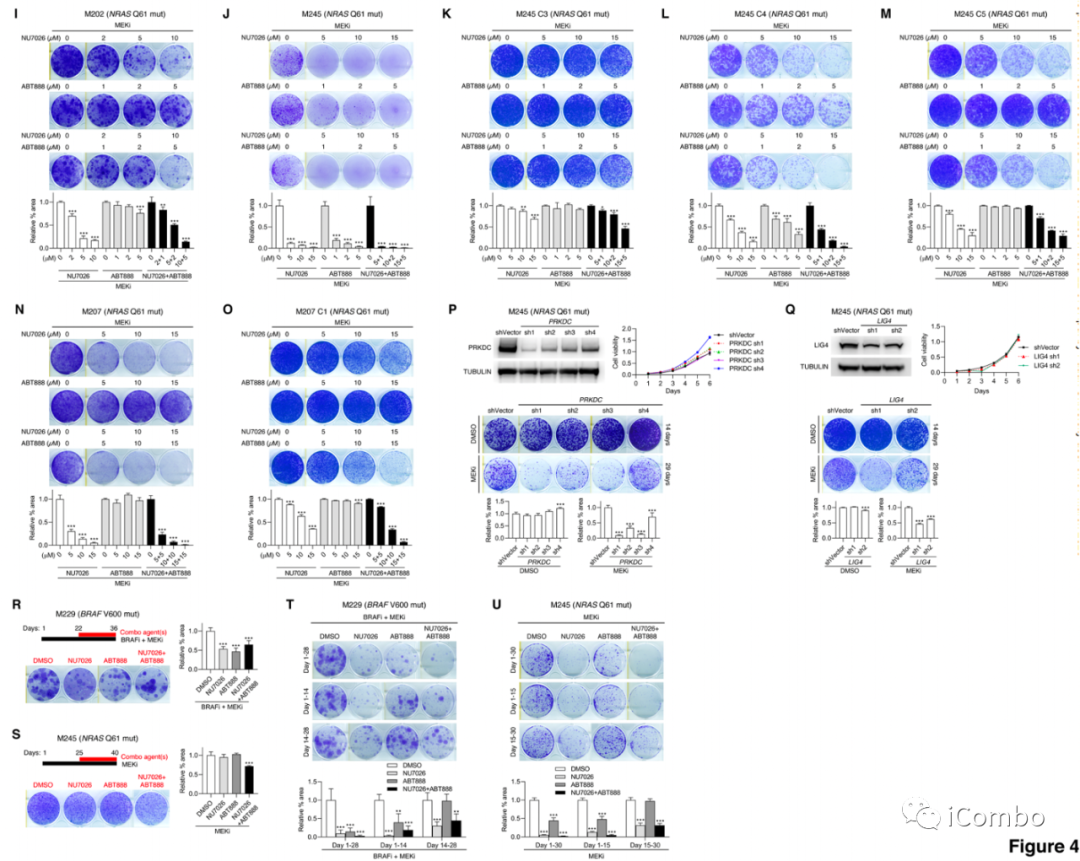
NU7026+MAPKi在所有亲本细胞系中具有协同作用和剂量依赖性,而ABT888在6个亲本细胞系中的3个中显示出活性(A/D/J)
A/D/J:在ABT888单独抑制DTPP形成的情况下,NU7026加ABT888对获得性MAPKi耐药的抑制作用甚至更强
在所有获得性MAPKi耐药的亚株中,与在等基因亲本株中观察到的活性相比,NU7026(或ABT888)在较低浓度范围内没有或降低了抗克隆活性
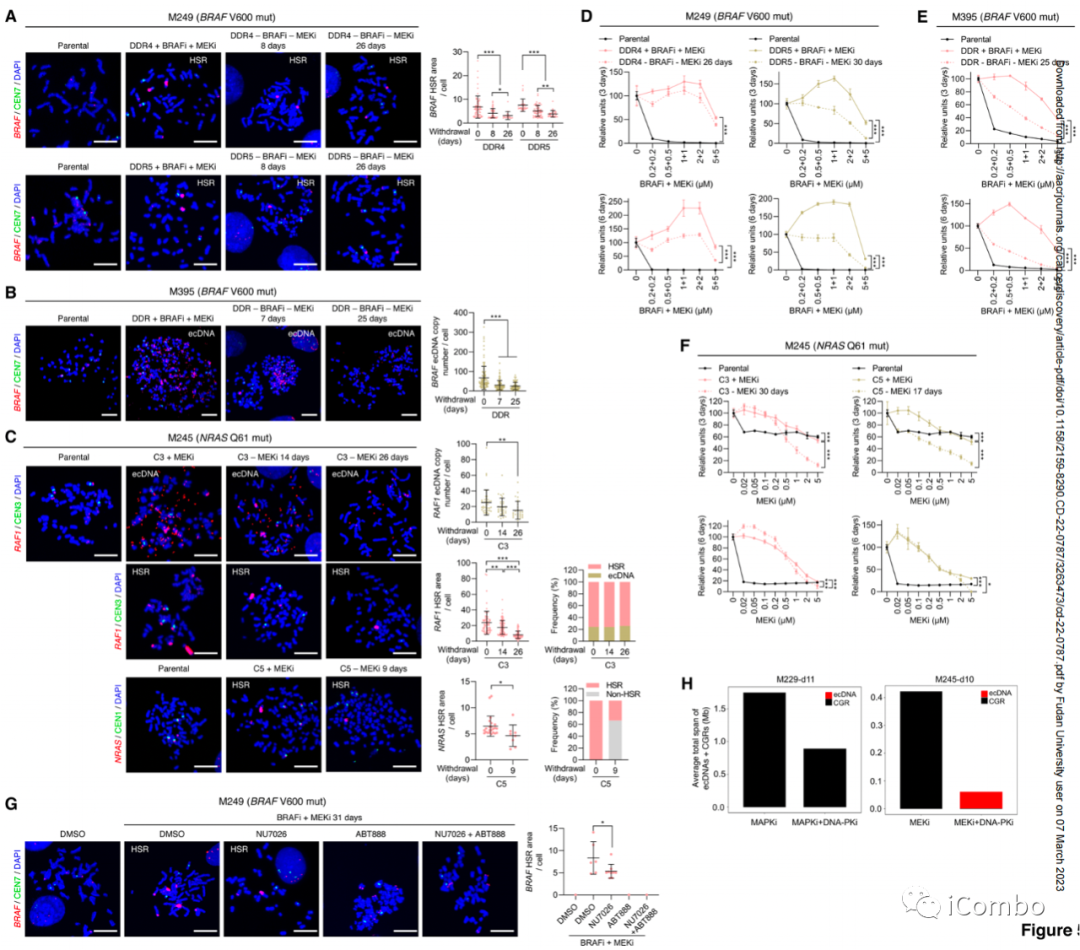
A~C:对中期细胞进行DNA-FISH,并对BRAF、RAF1和NRAS以及第7、3和1号染色体的着丝粒进行探测。在获得性耐药中观察到BRAF以HSRs(M249 DDR4 and DDR5)或ecDNAs(M395 DDR)的形式扩增;RAF1作为HSRs和ecDNA(M245 C3)的混合物;NRAS作为HSRs(M245 C5)。预期,这些驱动ecDNAs和HSRs在等基因亲本细胞系中未检测到。
A~F:观察到从获得性耐药亚系中撤出MAPKi显著减少了具有驱动ecDNAs和HSRs的中期细胞,并显著增强了它们的MAPKi敏感性
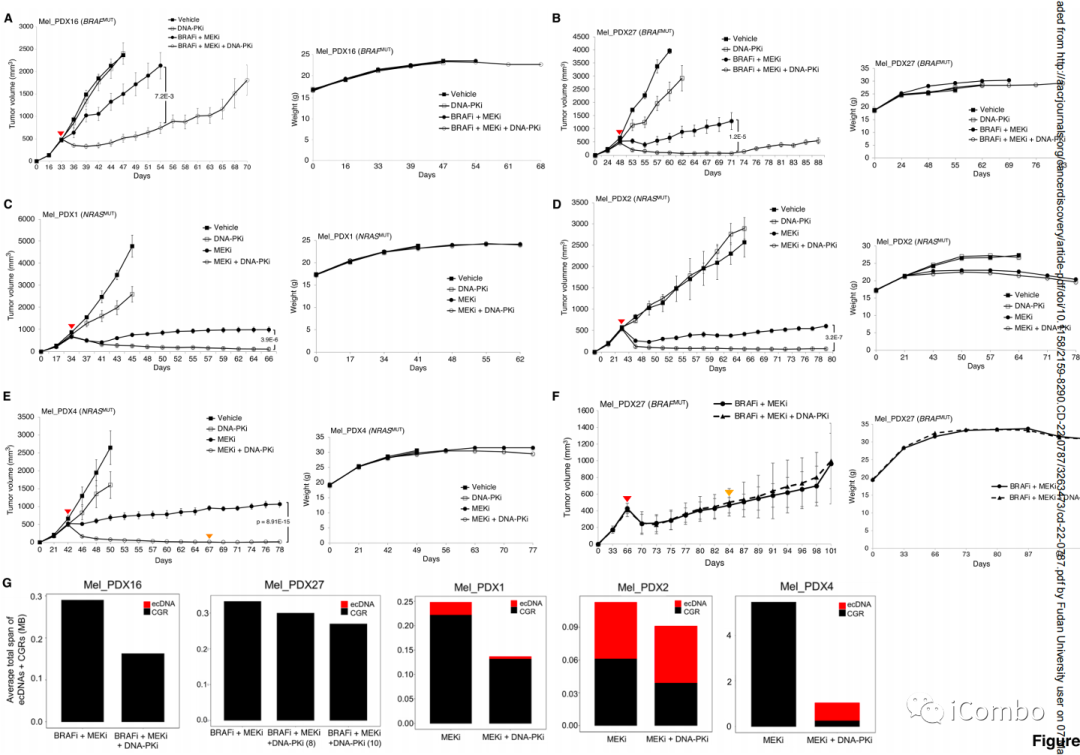
DNA-PKi在体内阻止耐药性并减少ecDNA和CGR大小
本网站所有内容来源注明为“williamhill asia 医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于williamhill asia 医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“williamhill asia 医学”。其它来源的文章系转载文章,或“williamhill asia 号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与williamhill asia 联系,williamhill asia 将立即进行删除处理。
在此留言









#黑色素瘤# #MAPK抑制剂#
65