专家论坛|杨永峰:自身免疫性肝炎诊治——病理必不可缺
2023-07-18 临床肝胆病杂志 临床肝胆病杂志 发表于上海

基于病理的诊断仍是AIH诊断金标准,因此病理检查在AIH的临床诊疗中必不可缺。
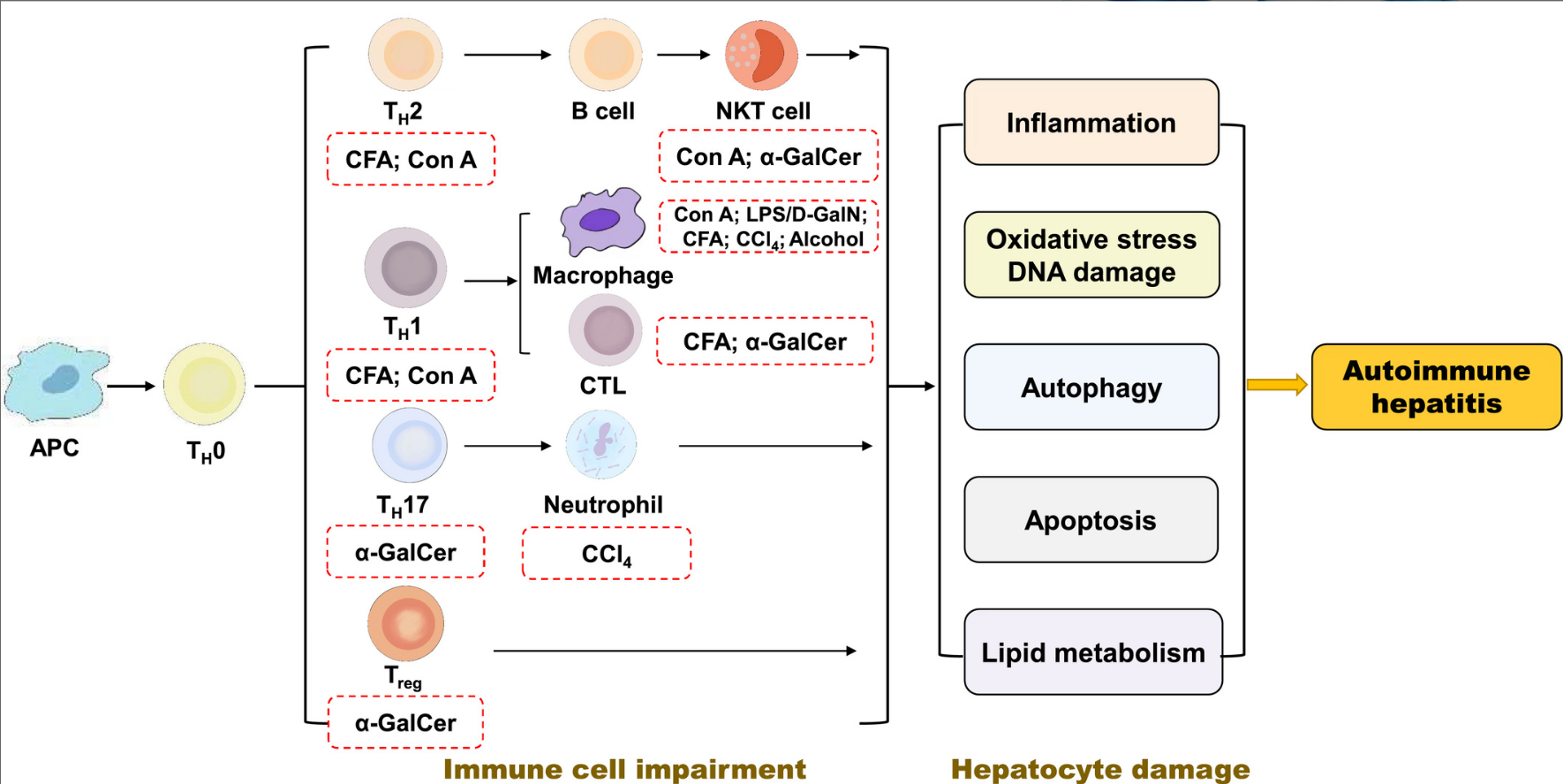
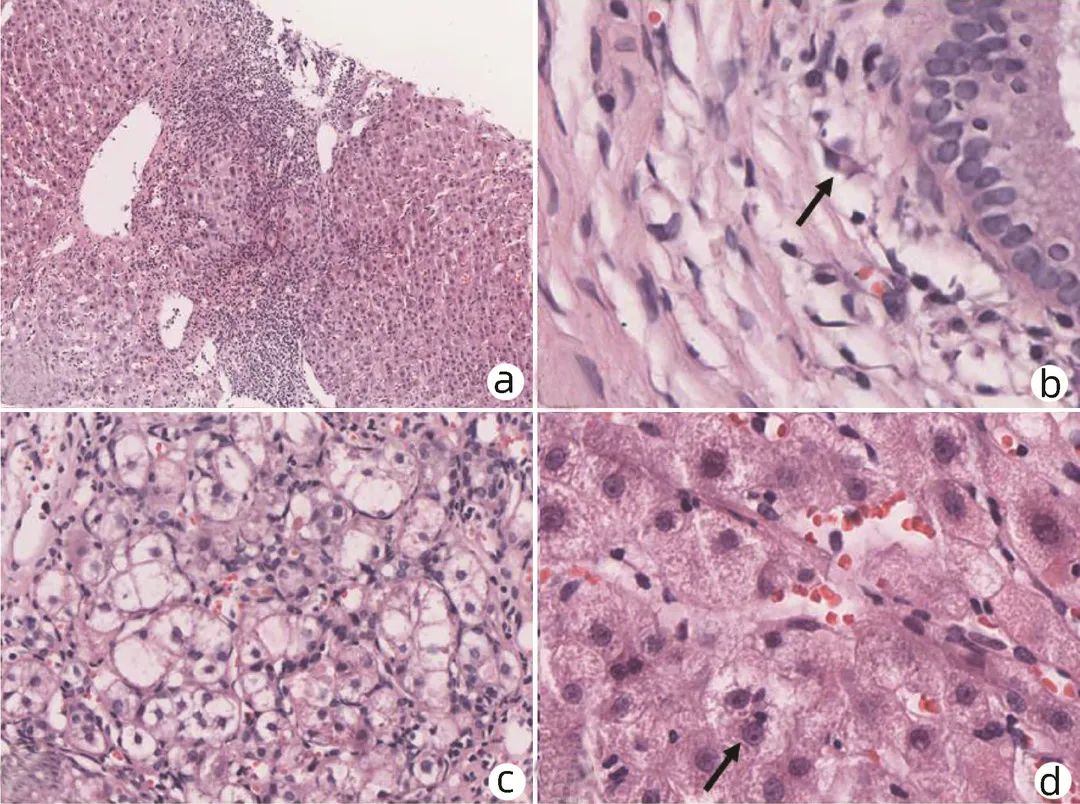
自身免疫性肝炎(AIH)可影响各年龄段所有种族患者,虽不同种族间存在差异,总体上,发病率正逐年增加。国内一项研究[1]显示:行肝穿刺活检的不明原因肝病患者中AIH占比19.1%,是不明原因肝病的主要构成之一。AIH常起病隐匿,或急性发病,部分患者确诊时已进入肝硬化阶段,最终可发展至肝衰竭期或致肝癌,直接威胁患者的生活质量及生命健康。目前,AIH的病因及发病机制尚未明朗,临床诊断难度较高,缺乏敏感性、特异性较高的标志物,疗效也存在一定个体差异。基于病理的诊断仍是AIH诊断金标准,因此病理检查在AIH的临床诊疗中必不可缺。 1、AIH临床特征 AIH是一种由异常自身免疫反应介导的针对肝细胞的肝脏实质性炎症病变,女性多发,部分患者合并肝外自身免疫性疾病。主要临床特征为反复发作的肝细胞损伤,伴或不伴典型临床症状,可自行缓解。血清学表现为氨基转氨酶水平升高、自身免疫抗体阳性、高免疫球蛋白血症(γ-球蛋白或IgG)等。 AIH多以慢性起病为主,可隐匿发病,部分患者在诊断时已进展至肝硬化阶段,少数患者表现为急性发作或肝衰竭起病。 (1)慢性发病AIH:患者表现为反复或持续发作的慢性肝炎,临床症状不典型且无特异性,症状多见乏力、纳差、黄疸、腹胀、脾大、皮肤瘙痒等,或表现为无症状隐匿起病,常常在体检中发现异常。 (2)急性发病AIH:具有急性肝炎的临床特征,相比较慢性起病,急性发作患者黄疸、疲劳、瘙痒、恶心、厌食等急性肝损伤症状发生率更高,往往伴高胆红素血症,炎症活动更严重,更容易进展为重型肝炎甚至肝衰竭,部分患者预后不佳[2]。且由于症状不典型、不具特异性,常被误诊为药物性肝损伤(DILI)或病毒性肝炎。 (3)AIH肝硬化:初期患者常隐匿进展,随着病情进展,部分患者有乏力、黄疸、肝脾肿大、皮肤瘙痒、体重下降等表现,随着纤维化进展、炎症活动减轻,临床和病理特征趋于不典型,至肝硬化失代偿阶段后,可出现腹水、肝性脑病、食管静脉曲张出血等重症[3]。 1.2.1 自身免疫抗体阳性 其中抗核抗体(ANA)和抗平滑肌抗体(ASMA)阳性最为常见,ANA和ASMA被认为是1型AIH(约占AIH病例的90%)的标记性抗体。Villalta等[4]研究示ANA的阳性检出率60%~70%、特异度约65%,SMA检出率约60%、特异度约95.5%,高达85%的AIH患者可检测到ANA和/或SMA。抗肝肾微粒体抗体-1型和抗肝细胞溶质抗原-1型是2型AIH(约占10%)的标记性抗体,诊断特异性较高,在2型AIH中抗肝肾微粒体抗体-1型检出率不低于70%,抗肝细胞溶质抗原-1型约30%。抗可溶性肝抗原抗体被认为是唯一的AIH特异性抗体,据统计特异度高达98.9%,但检出率只有20%~30%。英国一项多中心研究[5]结果示,在所有1267例AIH患者中,ANA检出率57%,ASMA检出率47%,抗肝肾微粒体抗体-1型仅测出2%,抗SLA阳性率24%。抗中性粒细胞胞浆抗体在1型AIH患者中约有30%阳性率,在原发性硬化性胆管炎患者中更常见[6-7]。 1.2.2 免疫球蛋白 IgG、γ-球蛋白升高是AIH特征性的血清免疫学改变之一,血清IgG水平可反映肝内炎症活动程度,经免疫抑制治疗后可逐渐恢复正常。故IgG不仅可作为AIH诊断的重要依据,对于监测疗效评价也有重要参考价值[8]。国内一项研究[9]显示,54.9%的AIH患者IgG水平可见升高,比例明显高于DILI组(17.9%)、慢性乙型肝炎组(45.9%),且AIH组患者血清IgG升高程度相比DILI、CHB组更明显。Gordon等[5]研究示78%的AIH患者可见血清IgG或球蛋白水平升高,尽管IgG增高对AIH有诊断价值,IgG正常并不能排除AIH诊断。IgG正常时,γ-球蛋白也可能升高,故同时检测这两项指标有助于提高检出率。部分患者基础IgG水平较低,疾病活动时即使IgG水平有所升高,也仍在正常范围内,但治疗后可见IgG水平下降。 1.2.3 肝功能(肝生化)指标特征 AIH主要表现肝细胞型肝损伤,血清ALT和AST水平反复不同程度的升高,多数患者可升至正常上限值的5~10倍。在Gordon等[5]研究中,ALT峰值水平为333(31~3785) U/L,AST为488(10~4181) U/L,首次检测ALT水平为182(13~3541) U/L,AST为255(10~3240) U/L。而血清ALP和GGT水平基本正常或轻微升高。在1999年版综合诊断积分系统中,ALP/ALT(或AST)的比值若>3即-2分,<1.5为+2分。 临床工作中需注意自身免疫抗体、免疫球蛋白是AIH的特征性表现但并非特异性标记,仅靠抗体阳性、免疫球蛋白升高不能直接诊断AIH,血清学阴性也不能排除AIH可能。另在疾病过程中免疫指标存在动态变化,早期自身抗体及免疫球蛋白呈阴性,建议隔3个月进行复查。 2、AIH病理特征 不同临床发病类型的AIH病理组织学表现不同,可表现急性炎症、慢性炎症、肝纤维化、活动性肝硬化等,总体以炎症型肝细胞损伤型改变为主。 2.1.1 界面性肝炎 肝细胞和汇管区/纤维间隔交界处称为“界板”。界板被炎细胞侵入,相邻肝细胞呈单个或小簇状坏死、脱落,炎症细胞沿破坏的界面向小叶内延伸并包绕坏死的肝细胞,称为界面性肝炎,如图1a所示。界面性肝炎是AIH的组织学特征性表现之一,中重度界面支持AIH诊断,但并非特异性,在其他慢性肝病中也同样存在,如药物性肝损伤、病毒性肝炎、Wilson病等。在Gordon等[5]研究中,88%的AIH患者出现界面炎,可见界面炎是AIH的特征性表现。 图1 慢性发病AIH病理特征(HE染色) 注:a, 界面性肝炎(×10);b, 浆细胞浸润,箭头示浆细胞(×40);c, 玫瑰花结(×40);d, 箭头示淋巴细胞穿入(×40)。 2.1.2 淋巴浆细胞浸润 汇管区及周围浸润的炎细胞主要为淋巴-浆细胞,如图1b所示。浆细胞主要见于汇管区,有时也可出现在小叶内,浆细胞评分>3分(浆细胞占炎症细胞≥20%)或小叶内/汇管区见浆细胞灶(≥5个浆细胞聚集为1灶)有助于AIH的诊断,大约有1/4确诊AIH患者浆细胞稀少甚至缺如,在Gordon等[5]的研究中淋巴-浆细胞浸润发生率约75%。AIH浆细胞主要为IgG阳性,少量为IgM阳性[10]。 2.1.3 玫瑰花结 肝细胞受炎细胞攻击后出现水肿、变性、坏死,数个水样变性的肝细胞呈假腺样排列,形似玫瑰花环而称为玫瑰花结样改变,周围可见淋巴细胞环绕,常见于界面炎周边,如图1c所示。 2.1.4 淋巴细胞穿入 指淋巴细胞进入肝细胞胞浆的组织学表现,多见于活动性界面炎,如图1d所示,是AIH的又一特征性表现。发生穿入的淋巴细胞主要为CD8+T淋巴细胞,可导致肝细胞凋亡。国内研究[10]表明,65%的AIH患者可见穿入现象,显著高于其他慢性肝病患者,并与AIH肝内炎症和纤维化程度相关。 急性发病AIH的表现与其他原因引起的急性肝炎类似,组织学可见小叶中央静脉周围炎症坏死(3区坏死)、桥接坏死,小叶内炎症细胞浸润,伴或不伴界面炎及汇管区炎症。伴有浆细胞浸润的小叶中心坏死被认为是急性发病AIH的特征性组织学表现。 日本一项多中心研究[11]评估了87例急性发病AIH患者,观察到多种形态的肝损伤类型:从轻度急性小叶炎(汇管区炎)到重型慢性肝炎,代表了急、慢性肝炎的整个组织学谱,临床可分为急性起病AIH(不伴纤维化)和慢性AIH的急性加重(具有不同活动度分级和纤维化分期)。其中84/86(97.7%)发生小叶炎症、坏死,75/84 (89.3%)发生淋巴细胞穿入现象,72/84 (85.7%)可见小叶浆细胞浸润,71/86 (82.6%)见鹅卵石样肝细胞,84.5%见蜡质样巨噬细胞,认为小叶炎症、坏死,小叶和/或汇管区浆细胞浸润,细胞穿入现象,蜡质样巨噬细胞,鹅卵石样肝细胞是急性期AIH最显著的特征。Aizawa等[12]通过评价113例AIH患者发现伴小叶中心区坏死的AIH相较于其他类型AIH,急性发作率较高,抗核抗体阳性率、滴度及IgG水平较低,界面炎程度较轻、淋巴浆细胞浸润不显著、AIH评分较低,推测小叶中心区坏死可能是急性发作AIH标志性组织学特征。目前,在国际自身免疫性肝炎小组(IAIHG)提出的修订版综合评分和简化评分系统中,并未纳入急性发作AIH特征,与急性发作的病毒性肝炎、DILI等表现相似,需注意鉴别诊断,避免误诊漏诊。 IAIHG于2022年发表了AIH病理诊断共识[20](表1),该共识将汇管区炎和小叶炎均作为AIH的病理特征,分别从汇管区炎症、小叶炎症的角度将肝组织学改变分为:极可能的、可能的、不可能的。主要关注的组织学特征包括汇管区及小叶内的界面炎、淋巴-浆细胞浸润等。该共识进一步优化了此前积分系统的不足,强调了病理诊断在AIH诊断中必不可缺的地位。 AIH可隐匿进展为肝硬化,伴随纤维化进展炎症会逐步消退,病理特征或不典型。此阶段界面炎程度较轻,表现为炎症活动程度不等的活动性肝硬化,部分病例仍可见浆细胞浸润、肝细胞玫瑰花结改变等。对于组织学表现不典型的病例需结合临床表现和治疗应答进一步鉴别。 3、AIH的临床病理诊断及鉴别诊断 AIH的基本临床特征为反复发作的肝细胞损伤,但其症状、表现均不典型,缺乏特异性标志物,诊断复杂,尚未有确定统一的金标准,需基于对临床表现、血清生化指标、免疫学指标和肝组织学表现的全面综合评估判断。AIH的诊断依据主要是在特征性病理表现(界面性肝炎、小叶炎、淋巴浆细胞浸润、玫瑰花结、穿入现象等)基础上,符合特征性临床表现,有反复发作的肝细胞型肝损伤,伴或不伴自身免疫抗体阳性、IgG升高,并排除其他可能的肝脏疾病。 在临床中遇到不明原因肝功能异常时需考虑AIH可能,首先详细追问疾病发展史及用药史,尽可能追溯实验室检查记录,其次检测肝功能、自身免疫抗体、免疫球蛋白等,对疑诊且无禁忌证的患者建议行肝穿刺活检。若组织学符合AIH特征性表现,再结合临床特征、实验室检查等并排除其他可能的肝脏疾病后进行AIH的综合诊断。如遇临床表现不典型、鉴别诊断困难的患者,可跟踪随访观察疾病的发展情况再予以明确诊断;诊断可能性较大且病情需要的患者,也可尝试予免疫抑制治疗后观察其免疫应答情况以辅助诊断。 AIH主要以慢性起病为主,主要病理特征是汇管区炎症、界面炎、局灶坏死等,与慢性病毒性肝炎、AIH样DILI、Wilson病等病理表现类似;少数患者呈急性发作,以小叶内炎症为主要表现,需与急性发作的病毒性肝炎、DILI等进行鉴别。 3.2.1 病毒性肝炎 包括常见的甲、乙、丙、丁及戊型肝炎,一般可通过血清病毒学指标鉴别病毒性肝炎。少数慢性丙型肝炎患者也可出现AIH的自身免疫表现,如自身抗体阳性、IgG升高等,同时也有极少数AIH患者可出现抗HCV假阳性反应出现,通过检测HCV RNA可予鉴别。组织学表现上,初诊的AIH较慢性丙型肝炎界面处及小叶内炎症更重,同时有些慢性乙型肝炎及少量丙型肝炎患者组织学可见浆细胞,另有大泡性脂肪变性多见于HCV(汇管区周围)和非酒精性脂肪性肝炎(小叶区),需注意鉴别[9]。 3.2.2 Wilson病 是由基因突变引起的铜代谢障碍的隐性遗传病,也可表现为慢性肝损伤,Wilson病组织学也可表现界面炎、淋巴浆细胞浸润、玫瑰花结、淋巴细胞穿入等,可和AIH重叠。Wilson病常见于年轻患者,主要通过肝组织铜沉积、铜蓝蛋白、血清和尿铜浓度及基因的检测予以区分,裂隙灯检查出现K-F环也是Wilson病的重要诊断依据。 3.2.3 DILI DILI以急性发病为主,少数也可慢性隐匿起病,按临床特征分为肝细胞损伤型、胆汁淤积型或者混合型[13]。部分患者也会出现自身免疫抗体阳性、IgG升高等,缺乏特异性的标志物,尤其是AIH样DILI(免疫介导的DILI、AL-DILI)、药物诱导的AIH(DI-AIH)及AIH基础上合并的DILI[14]临床表现相似度高,鉴别诊断困难。(1)临床中遇到反复发作的DILI,需考虑AIH基础上合并DILI的可能,组织学常见进展性纤维化[15];(2)DI-AIH是由药物诱发,但其本质为AIH,对免疫抑制治疗应答良好,需要长期治疗;(3)AL-DILI多见于肝细胞损伤型和混合型患者中,组织学表现类似于AIH,可见汇管区中性粒细胞浸润及胆汁淤积,停用药物后改善不显著,但对激素治疗应答良好,与AIH不同的是,AI-DILI患者不会出现反复发作的肝损伤。 DILI和AIH都可表现为肝细胞型损伤,AIH以慢性肝炎为主反复发作,只有约10%呈急性发作,而DILI[16]以急性发病为主,长期用药也可呈慢性肝损伤,停用致损伤药物后病情好转。临床上遇到这类患者,首先仔细询问用药史,判断有无可疑药物,其次完善实验室相关检查,DILI目前最常用RUCAM评分辅助诊断,最后诊断不明的患者可行肝穿刺活检,结合病理诊断综合评价有助鉴别,帮助临床决策,避免误诊。 IAIHG于1999年修订了自身免疫性肝炎的综合诊断评分[17],并于2008年发布简化诊断评分[18]。 综合评分主要内容包括临床特征7分、实验室检查14分、组织病理学5分、治疗应答3分,根据是否接受治疗分为治疗前、治疗后,治疗前确诊需>15分,10~15分为可能诊断,治疗后确诊需>17分,12~17分为可能诊断。肝组织学具体如下:界面性肝炎+3、门管区和小叶内淋巴-浆细胞浸润+1、肝细胞呈玫瑰花结样改变+1、无上述表现-5、胆管改变-3、其他非典型改变-3。综合积分系统对于诊断AIH有良好的敏感性和特异性,但过于复杂,难以在临床实践中全面推广应用。且部分指标如HLA-DR或DR4在国内开展检测较少,仅仅靠评分标准容易出现漏诊。需要强调的是,综合评分中支持AIH的组织学表现为加分项,不支持AIH和支持其他疾病的组织学表现为减分项。临床实践中有未行组织学检查时综合评分达到“可能诊断”甚至是“确定诊断”,组织学检查后因组织学不支持而减分,从而否定诊断的病例并不少见,故强调评分是有组织病理学结果前提下的评分,无组织学检查结果时慎用评分。 简化诊断评分[18]主要包括自身抗体、血清IgG水平、肝组织学改变和排除病毒性肝炎4个部分。AIH主要特征性表现包括界面性肝炎, 门管区/小叶内淋巴-浆细胞浸润、肝细胞玫瑰花结样改变及细胞穿入现象。肝组织学变化归纳为三类:“典型”AIH为4项中满足3项得2分;“符合”AIH即有淋巴细胞浸润的慢性肝炎表现,但不足3项特征性表现,得1分;“不典型”则有支持其他诊断的组织学依据,得0分。诊断标准总分8分,确诊需≥7分。无组织学结果前提下,即使其他结果都符合,简化评分也只能达到6分的可能诊断,不能达到确定诊断。 国内一项样本量为405例的多中心研究[19]包含了1型AIH、PBC、AIH - PBC重叠、DILI、非酒精性脂肪性肝炎、CHB、慢性丙型肝炎等慢性肝病病例,结果示简化评分和综合评分诊断“可能的AIH”敏感度分别为90%和100%、特异度为95%和93%;诊断“确定的AIH”敏感度分别为62%和64%,特异度为99%和100%;认为综合评分和简化评分均可用于国内AIH病理诊断,同时发现简化评分容易漏诊部分不典型患者,如自身抗体滴度低或阴性和/或血清IgG水平较低甚至正常的患者。现简化标准已成为临床中首选评分标准,对于表现不典型的患者再选用综合评分进行评估。与综合评分相比,简化评分诊断实用性更好,其中组织学表现占比提高,进一步强调了肝组织学检查在AIH诊断中必不可缺的作用。 虽然这两种评分诊断效能良好,但在临床实际应用中仍存在一定不足。如两种评分标准主要关注慢性AIH,其炎症改变主要发生在汇管区。急性AIH炎症改变通常发生在小叶内,两种评分均未纳入该病理表现,对这部分病例诊断建议参考IAIHG的诊断共识,在排除急性发作的病毒性肝炎、DILI等类似临床表现疾病前提下做出诊断。 4、组织病理学在AIH诊治中作用 AIH诊断缺乏金标准,其特征性临床表现及血清学指标如自身抗体(ANA、ASMA等)、IgG等均缺乏特异度,敏感度也欠佳。因此,脱离病理的临床诊断是不可靠的,完善病理组织学检查有助于明确诊断、评估病情、判断预后,帮助临床决策。病理在AIH的诊疗过程中是必不可缺的,但组织病理学也并非“金标准”,病理和临床紧密结合才能实现精准诊断。 AIH目前的诊断现状[21]是诊断不足与诊断过度共存,诊断不足主要原因是: (1)对于AIH的临床特征认识不足,尤其是急性发作的AIH、AIH肝硬化及AIH肝衰竭等临床特征不典型的类型,往往会忽视AIH可能; (2)仅凭免疫学指标正常,未行病理检查即排除AIH诊断; (3)仅关注病理组织学表现而未结合临床,或不了解AIH的非典型病理改变,而不予诊断AIH。诊断过度主要原因是:(1)未行肝穿刺活检,仅依靠免疫学指标异常和/或肝功能异常即予以诊断;(2)仅依靠组织学表现,不重视临床病史,即建立诊断。AIH的诊断需结合临床与病理,缺一不可。 AIH应与DILI、病毒性肝炎、遗传代谢性肝病等多种疾病鉴别,对于靠临床表现难以鉴别的病理,结合组织学特征鉴别诊断至关重要。 AIH的明确诊断需在病理特征支持的基础上,符合相应临床特征(反复发作的肝细胞型肝损伤,伴或不伴IgG增高、自身抗体阳性),并排除其他临床及病理表现类似的疾病。在病理基础上可借助评分标准,但不能唯评分是论。病理学在AIH的诊断中是必不可缺的。 从肝组织学角度,中度以上界面性肝炎即是治疗的重要指征,若有急性AIH或重症表现,需及时启动免疫抑制治疗。表现为轻度界面炎者,年轻相对较轻、症状明显的患者有进展至肝硬化的风险,可酌情启动治疗,年老及有治疗禁忌证者则严密观察、暂缓用药。 AIH的整体治疗目标是获得并维持肝组织学缓解、防止疾病进展,提高患者的生活质量和生存期。生化缓解定义为ALT、AST及IgG水平恢复正常;组织学缓解定义为肝内轻微炎症或炎症消失(Ishak评分HAI<4或Scheuer分级G≤1)[23]。肝组织学炎症的消退往往迟于生化缓解,因此对组织学的应答评估可能需要延后6~12个月。患者获得生化缓解2年以上方可考虑停药,而停药前进行肝组织学复查是很重要的,组织学上界面炎和小叶内炎症的减退或消失可定义为疾病缓解,部分患者还可出现纤维化逆转的情况,界面炎伴浆细胞存在时停药可致疾病复发,达到组织学缓解者复发率可降低[24]。 5、总结和展望 尽管组织病理学是有创检查,但其对AIH的诊断必不可缺,对指导治疗和停药也至关重要,在AIH诊断和治疗过程中要重视临床和病理的结合。未来需要更多的研究,开发敏感性、特异性能满足临床需求的诊断和预后指标,用于指导AIH诊断和治疗。 王丽, 杨永峰. 自身免疫性肝炎诊治——病理必不可缺[J]. 临床肝胆病杂志, 2023, 39(3): 504-510. 1.1 临床表现
1.2 实验室指标特征
2.1 慢性发病AIH
2.2 急性发病AIH

2.3 AIH肝硬化
3.1 AIH的诊断
3.2 鉴别诊断
3.3 诊断评分
4.1 诊断和鉴别诊断
4.2 指导治疗
本网站所有内容来源注明为“williamhill asia 医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于williamhill asia 医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“williamhill asia 医学”。其它来源的文章系转载文章,或“williamhill asia 号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与williamhill asia 联系,williamhill asia 将立即进行删除处理。
在此留言